
Production d’amines aromatiques
Les amines aromatiques sont produites par trois types de réactions :
- Réductions : en utilisant des éléments métalliques comme le fer (Fe), le zinc (Zn), l’étain (Sn), l’aluminium (Al) ou leurs sels correspondants ; composés soufrés; procédés électrochimiques; et l’hydrogénation catalytique.
- Substitutions nucléophiles : impliquant l’échange de substituants tels que les groupes halogène, hydroxyle, alcoxy et sulfonique.
- Réarrangements et dégradations : y compris des transformations telles que les réarrangements de la benzidine et de Beckmann, ainsi que les dégradations de Schmidt et Hofmann.
Il convient de noter que les deux premiers types de réaction sont plus importants. Les réarrangements chimiques et les dégradations aboutissent rarement à des produits de réaction purs avec des rendements élevés.
Table des matières
1. Production d’amines aromatiques par réduction de composés nitrés
Les amines aromatiques sont produites par réduction de composés aromatiques carbone-azote contenant des états d’oxydation de l’azote allant de +3 à +2 can. Notamment, la réduction des composés nitrés a été largement utilisée à l’échelle industrielle en raison de la préparation précise des matières premières qui peut être obtenue à travers une gamme variée de composés.
Cependant, la faisabilité économique de la production d’autres composés azotés aromatiques en tant que matières premières viables pour les amines aromatiques est limitée, limitant ainsi leur utilité pratique.
1.1. Réduction chimique
Au cours du processus de réduction chimique qui convertit les composés nitrés aromatiques en amines aromatiques, l’atome d’hydrogène qui se lie à l’atome d’azote provient généralement du solvant, souvent de l’eau, ou est introduit par l’utilisation d’acide ajouté.
Parmi les agents essentiels de la réduction, divers métaux comme le fer, l’étain et le zinc ont la priorité, ainsi que le phosphore, les sulfures, les sulfites et le dioxyde de soufre, qui sont également utilisés.
Il est pertinent de noter que l’oxydation potentielle de l’agent réducteur, conduisant à la formation d’un déchet potentiellement inutilisable, nécessite des mesures d’élimination respectueuses de l’environnement.
En conséquence, l’importance de la réduction chimique a diminué par rapport à la réduction catalytique, en particulier dans la synthèse à grande échelle de divers produits.
1.1.1. Réduction au fer
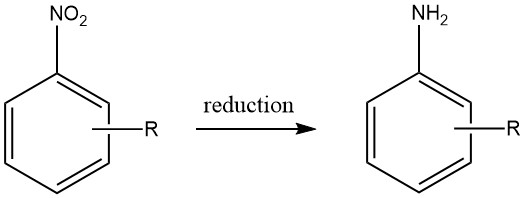
Le procédé de réduction des composés par le fer aussi appelé réduction de BÉCHAMP a été initialement élucidé en 1854 et est souvent réalisé en milieu acide dilué, . Le fer conserve son rôle prééminent en tant que catalyseur métallique primaire pour la conversion des composés nitrés en leurs amines aromatiques correspondantes.
Pratiquement tous les composés nitrés peuvent être réduits avec succès en amines aromatiques en utilisant du fer dans de l’eau et un milieu acide. Notamment, l’émergence de sous-produits indésirables est principalement anticipée lorsque la molécule contient d’autres substituants susceptibles d’être réduits, tels que des groupes nitroso, azoïque, hydrazine, sulfoxyde ou nitro supplémentaires, ou si elle peut subir une saponification.
Les liaisons multiples carbone-carbone restent inertes à l’attaque. En évidence, Bayer (Allemagne) et Mobay (États-Unis) mettent en œuvre l’approche de réduction médiée par le fer pour générer des oxydes de fer, produisant de l’aniline comme sous-produit.
Un autre cas d’importance industrielle est l’emploi exclusif de fer en milieu neutre pour réduire l’acide 4,4′-dinitro-2,2′-stilbènedisulfonique en acide 4,4′-diamino-2,2′-stilbènedisulfonique. De plus, de nombreux intermédiaires de colorants de volume limité subissent une réduction via la méthode à médiation par le fer.
1.1.2. Réduction avec d’autres métaux
Les groupes nitro aromatiques subissent une réduction en utilisant du zinc, de l’étain et de l’aluminium, ou leurs sels correspondants, en milieu acide, neutre ou alcalin.
Ces agents réducteurs sont doux et n’interfèrent donc pas avec les groupes fonctionnels tels que -OH, -OR, -COOH, -CO-Ar, halogène ou -CN. Bien qu’ils trouvent des applications dans des contextes de laboratoire, leur utilisation pratique manque d’importance industrielle substantielle.
Ceci est principalement attribué à l’utilisation répandue de l’hydrogénation catalytique, qui s’aligne sur les directives réglementaires concernant les pratiques de gestion et d’élimination des eaux usées.
1.1.3. Réductions avec du sulfure, de l’hydrogénosulfite et du dioxyde de soufre
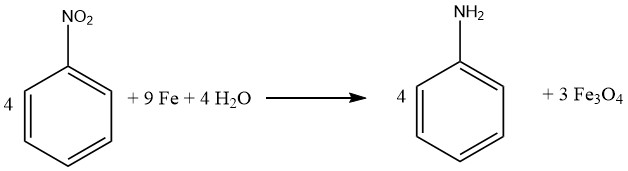
Les composés contenant plusieurs groupes nitro peuvent être réduits, produisant des nitro amines, en utilisant une quantité équimolaire de réactif sulfure. Le sulfure de sodium ou d’ammonium est préféré. Un rendement de 87 % de m-nitroaniline est obtenu en réduisant le 1,3-dinitrobenzène avec du sulfure de sodium.
L’ajout de soufre élémentaire diminue la quantité requise de sulfure de sodium. Par exemple, le 4-nitrotoluène est réduit avec une grande efficacité pour donner du 4-aminobenzaldéhyde en utilisant du sulfure de sodium et du soufre.
Les groupes azoïques restent inchangés lorsque la réduction utilisant du sulfure de sodium se produit dans la plage de température de 40 à 70 ° C, avec de courtes durées de réaction et en l’absence d’excès de sulfure.
Cependant, les substituants halogène sont facilement substitués par des groupes -SH. Dans les cas où un excès d’agent réducteur n’est pas un problème, l’hydrogène sulfuré d’ammonium en présence d’ammoniac peut être utilisé.
La réduction des composés aromatiques nitro, nitroso ou azoïques en leurs amines aromatiques correspondantes à l’aide de sulfite ou d’hydrogénosulfite est reconnue comme la réaction de Piria (PIRIA, 1851).
Dans des conditions aqueuses ou alcooliques, principalement des acides N- et C-sulfoniques sont générés. Par traitement avec un acide minéral, un mélange d’amines aromatiques et d’acides aminobenzènesulfoniques se forme.
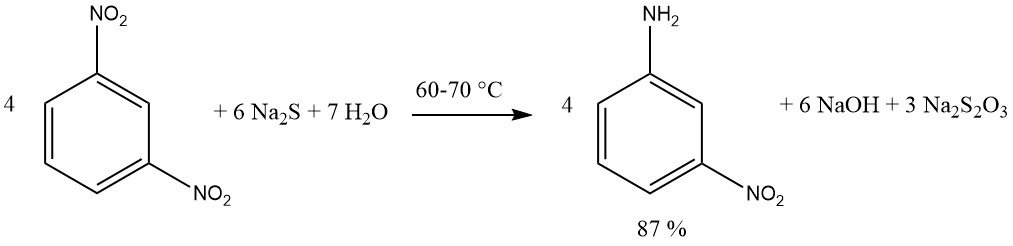
La présence d’un intermédiaire quinone oxime facilite l’introduction d’hydrogénosulfite sur le cycle aromatique lors de la réduction des ortho- et para-nitroso-phénols ou des naphtols.
Plus précisément, lorsque le groupe hydroxyle est en position para, le groupe sulfo sera incorporé en position 2. Les substituants positionnés en position 3, tels que Cl ou COOH, sont remplacés par l’hydrogène.
L’utilisation industrielle de la réduction avec du dioxyde de soufre (SO2) n’est devenue pertinente que lorsque la réaction secondaire de C-sulfonation a été réduite.
Cette procédure est menée de manière optimale dans un système scellé, en utilisant des conditions fortement acides (par exemple, 15 à 40 % d’acide sulfurique aqueux) et des températures allant de 80 à 180 °C. Les catalyseurs tels que l’iode, l’iodure d’hydrogène ou les sels d’iode font partie intégrante, et les réacteurs fabriqués à partir de matériaux émaillés sont optimaux.
Le dithionite de sodium produit une réduction presque quantitative des groupes aromatiques nitro et nitroso.
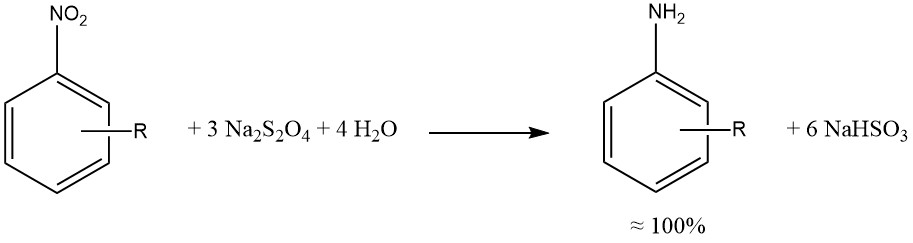
Dans la mesure du possible, les réductions par le sulfure, le sulfite et d’autres composés soufrés ont été remplacées par des hydrogénations catalytiques, produisant des mélanges réactionnels plus propres, une corrosion réduite (associée à l’utilisation de SO2) et moins de problèmes environnementaux.
1.1.4. Réduction électrochimique des composés nitrés
La réduction électrochimique est généralement considérée comme une variante spécialisée de la réduction chimique. Dans cette approche, un composé inorganique sert d’agent réducteur, et la forme oxydée est ensuite réduite à la cathode, lui permettant de réagir à nouveau.
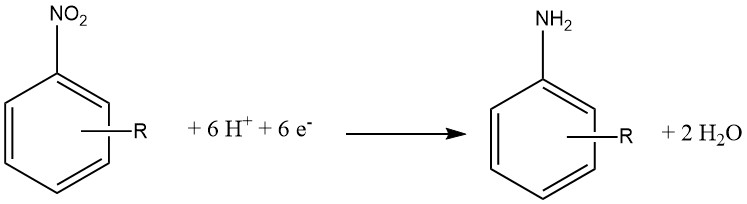
Le matériau de la cathode peut être constitué de Pb, Sn, Ni ou Cu et de l’acide chlorhydrique à 15 à 20 % est utilisé du côté cathode d’une membrane semi-perméable. Côté anode, on utilise de l’acide sulfurique à 30 %.
Bien que les réductions électrochimiques soient connues depuis plus d’un siècle, elles n’étaient pas largement utilisées dans les applications commerciales jusqu’à récemment. Cependant, l’intérêt pour cette méthode augmente à mesure que les progrès de la technologie cellulaire et des membranes continuent de s’améliorer.
Certains produits ont été produits avec succès par réduction électrochimique, en particulier en Inde, comme l’acide p-aminobenzoïque, le p-aminophénol et plusieurs autres, qui ont atteint l’échelle de l’usine pilote ou au-delà.
1.2. Hydrogénation Catalytique des Composés Nitrés
1.2.1. Réduction à l’hydrazine
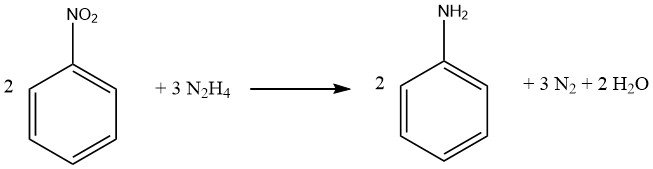
Le processus de réduction des composés nitrés aromatiques à l’aide d’hydrazine est une forme spécialisée d’hydrogénation catalytique, dans laquelle l’hydrazine sert de source d’hydrogène.
Le mécanisme de décomposition de l’hydrazine sur les catalyseurs de métaux précieux varie en fonction de la valeur du pH, ce qui entraîne une augmentation du rendement en hydrogène par mole d’hydrazine à des niveaux de pH plus élevés.
Dans des conditions faiblement alcalines ou neutres, 1 mole de H2 est produite.
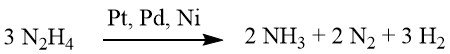
Cependant, lorsqu’il est complété par de l’hydroxyde de baryum ou du carbonate de calcium, ce rendement grimpe à 2 moles de H2.
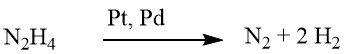
Bien que la réduction à l’aide d’hydrazine puisse parfois se dérouler sans catalyseur, elle est généralement réalisée en présence d’un catalyseur d’hydrogénation, tel que le nickel de Raney, le palladium sur charbon ou le palladium sur carbonate de calcium.
Idéalement, 1 mole d’hydrazine fournit quatre équivalents de réduction, en fonction du pH et du catalyseur, bien que moins d’équivalents soient pratiquement générés, ce qui nécessite un excès d’hydrazine. Il est important de noter que les doubles liaisons carbone-carbone et les groupes carbonyle restent inchangés. L’isolement des intermédiaires au cours du processus de réduction dépend des conditions de réaction.
Dans les cas de réduction non catalytique avec de l’hydrazine, les composés contenant des groupes carbonyle sont réduits en leurs composés hydroxy correspondants via la formation d’intermédiaires hydrazone.
La réduction de composés nitrés aromatiques à l’aide d’hydrazine, par rapport à la réduction catalytique utilisant de l’hydrogène, présente des avantages limités. Il trouve son utilité principalement dans les expériences de laboratoire et pour les petits volumes de produits, où des conditions de pression normales peuvent être utilisées.
À l’échelle du laboratoire, le cyclohexène a également été exploré comme source d’hydrogène pour l’hydrogénation catalytique de composés polynitro.
1.2.2. Réduction des groupes nitro avec l’hydrogène
La génération de mono- ou polyamines aromatiques primaires implique généralement une hydrogénation catalytique du composé nitré correspondant, soit conduite en phase vapeur, soit en phase liquide, et peut être exécutée avec ou sans la présence d’un solvant. De nombreux produits importants, notamment l’aniline, l’o- et p-toluidine, le 2,4- et 2,6-diaminotoluène et la 1-naphtylamine, sont fabriqués à l’aide de cette méthode.
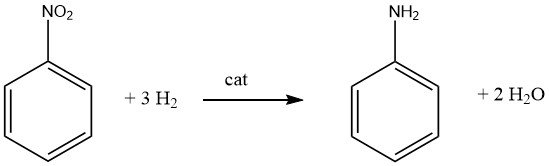
Il est important de reconnaître que la réduction catalytique des composés nitrés est exothermique. Lors de l’hydrogénation en phase liquide du nitrobenzène, une libération de 553,5 kJ/mol d’énergie se produit, tandis que l’hydrogénation en phase vapeur à 200 °C libère 493,2 kJ/mol.
Si elle n’est pas suffisamment dissipée, cette chaleur peut conduire à des explosions potentiellement dangereuses, en particulier si la décomposition thermique du composé nitré se produit ou si des réactions de condensation se déclenchent, comme dans le cas des composés chloro-nitro.
Pour pallier ces risques, la concentration du composé nitré, la quantité et la pression partielle d’hydrogène, la température et l’activité du catalyseur sont minutieusement régulées.
Hydrogénation en phase vapeur
L’application industrielle de l’hydrogénation en phase vapeur est limitée par le point d’ébullition et la stabilité thermique des composés nitrés.
Aux États-Unis, une partie importante de l’aniline est produite par hydrogénation en phase vapeur du nitrobenzène à l’aide d’un catalyseur cuivre-silice, ce qui donne un taux de conversion impressionnant de 99 %. Bayer utilise de l’alumine Pd à cette fin en Europe et au Brésil.
Hydrogénation en phase liquide
L’hydrogénation en phase liquide de la plupart des composés nitrés aromatiques est généralement préférée. Dans ce scénario, la pression et la température peuvent être manipulées indépendamment. Il est important de noter que des températures supérieures à 170–200 °C peuvent favoriser les réactions d’hydrogénation impliquant le cycle aromatique.
En tant que tel, le processus de réduction est généralement effectué dans la plage de 100 à 170 ° C. Certains composés, tels que le 1-bromo-4-nitrobenzène, nécessitent des températures encore plus basses (20 à 70 ° C) pour empêcher le clivage des groupes sensibles.
Industriellement, des pressions allant de 1 à 15 MPa (10 à 150 bars) sont utilisées. Pour l’hydrogénation de composés nitroaromatiques avec une sensibilité accrue, des pressions plus basses (0,1–5 MPa, 1–50 bar) sont recommandées.
La pression affecte principalement la vitesse de réaction en influençant le transfert de phase et la saturation du catalyseur en hydrogène. Le temps de réaction dépend de divers paramètres, notamment la pression d’hydrogène, la concentration, la température, l’activité et la concentration du catalyseur et le mélange.
Souvent, une période d’induction initiale est observée, qui n’est pas affectée par l’activité du catalyseur. Les temps de réaction s’étendent généralement de quelques minutes à plusieurs heures. Des durées plus longues sont nécessaires si ces paramètres ne sont pas convenablement optimisés.
En raison de la nature intrinsèquement exothermique de la réaction, des précautions de sécurité strictes doivent être respectées, en particulier dans l’hydrogénation industrielle de composés polynitro aromatiques en l’absence de solvants.
Le caractère exothermique est géré par l’ajout continu de petites quantités du composé nitré, en maintenant sa concentration inférieure à 2 %. De l’eau déionisée peut être introduite pour absorber la chaleur de réaction par évaporation continue.
Cela atténue non seulement les problèmes liés à la chaleur, mais affecte également l’activité du catalyseur ; une teneur en eau plus élevée dans l’hydrogénation discontinue du dinitrotoluène sans solvant conduit à une activité réduite du catalyseur Pd sur carbone.
Si l’amine est soluble dans l’eau, elle peut être utilisée comme solvant. Cette approche convient également lorsque le composé nitré forme des sels solubles dans l’eau avec un alcali, comme on le voit avec les acides nitrocarboniques ou sulfoniques.
Dans certains cas, des solutions de l’amine dans l’eau (30 à 40%) peuvent être directement générées dans le réacteur d’hydrogénation, évitant ainsi la nécessité d’étapes de concentration supplémentaires.
Les solvants, tels que le méthanol et le 2-propanol, sont généralement privilégiés. Le dioxane, le tétrahydrofurane et la N-méthylpyrrolidone ont également été utilisés. Par exemple, l’o-nitrophénol atteint de bons rendements d’hydrogénation dans le méthanol en utilisant du Pt sur du carbone.
Lors de l’utilisation d’un solvant non miscible à l’eau, tel que le toluène, la teneur en eau doit être minimisée pour préserver l’activité du catalyseur, similaire à l’hydrogénation sans solvant. Dans certains cas, des composés nitrés hautement polaires, comme la bis(4-amino-3-nitrophényl)sulfone, sont hydrogénés à l’aide d’ammoniac liquide.
Catalyseurs d’hydrogénation
L’hydrogénation en phase vapeur repose sur des métaux ou des dérivés métalliques sur des supports dans des lits fixes ou des lits fluidisés. L’hydrogénation en phase liquide utilise principalement des métaux avec des surfaces étendues, tels que le nickel de Raney, le nickel-fer de Raney, le cobalt de Raney et le cuivre de Raney, en raison de leur coût relativement inférieur.
Les catalyseurs à base de métaux précieux, tels que Pt et Pd, sont généralement utilisés à des concentrations de 0,5 à 5 % en poids sur des matériaux de support à surface élevée tels que le charbon de bois, la silice, l’oxyde d’aluminium ou les carbonates alcalino-terreux.
La forme de catalyseur en poudre convient aux boues, tandis que les formes de pastilles sont utilisées dans des lits fixes. Il est impératif d’optimiser les conditions permettant la réutilisation et la régénération du catalyseur, tout en étant économique pour une production à grande échelle (> 500 t/a).
Les sous-produits de décomposition et d’oxydation conduisent fréquemment à la désactivation du catalyseur par blocage de surface. Même de petites quantités de composés de soufre, d’arsenic ou d’antimoine peuvent empoisonner les catalyseurs. Cependant, ces poisons de catalyseur présentent moins d’impact à mesure que les états d’oxydation augmentent (par exemple, As3- → As5+ or S2- → S4+).
Avec les catalyseurs de métaux nobles, les ions halogénure, sodium, magnésium, cobalt et CO2 peuvent réduire l’activité. Les composés nitroaromatiques contenant du soufre présentent une meilleure réduction en utilisant du sulfure de molybdène ou de tungstène, ou avec des catalyseurs au nickel contenant du zinc ou du carbonate de calcium.
Les catalyseurs Pd sur carbone peuvent être affectés par les sels de cuivre, tandis que le nitrophénol et le nitrocrésol agissent comme de puissants poisons de catalyseur et activateurs de décomposition lors de l’hydrogénation du dinitrotoluène.
Hydrogénation de composés nitrés à substitution halogénure
L’hydrogénation de composés nitrés avec des substitutions d’halogénure nécessite souvent des températures plus basses pour empêcher l’échange halogénure-hydrogène. Les catalyseurs, en particulier les catalyseurs métalliques empoisonnés au sulfure, présentent une activité réduite dans de telles conditions.
Le 1-chloro-2-nitrobenzène peut être efficacement réduit à 80 °C et 1,0–5,0 MPa (10–50 bar) à l’aide d’un catalyseur à 5 % en poids de Pt sur carbone traité avec de l’acide sulfurique dilué, suivi de H2 et H2S. Ce catalyseur peut être recyclé jusqu’à 30 fois sans purification supplémentaire.
Des méthodologies similaires ont été employées pour l’hydrogénation du 3-chloronitrobenzène en utilisant des catalyseurs traités avec du thiocyanate. L’utilisation de catalyseurs modifiés améliore la sélectivité et l’efficacité de la réaction, ce qui se traduit par des rendements élevés et une excellente pureté du produit.
Équipement et matériaux de construction
L’hydrogénation en phase vapeur est généralement réalisée en mode continu, tandis que l’hydrogénation en phase liquide peut être réalisée par des procédés discontinus ou continus, selon l’échelle. L’hydrogénation continue en phase liquide est principalement utilisée pour quelques produits sélectionnés à grande échelle, tels que le dinitrotoluène, le nitrotoluène et le 1-nitronaphtalène.
Cependant, la majorité de la production d’amines aromatiques implique une hydrogénation discontinue à l’aide de suspensions de catalyseur. Cette approche permet des transitions rapides entre les produits, un luxe qui n’est pas offert lors de l’utilisation de catalyseurs à lit fixe.
Un mélange efficace des trois phases (hydrogène gazeux, solution de composé nitré et catalyseur solide) est important. Le transport rapide des réactifs vers les centres catalytiques actifs et l’élimination ultérieure du produit sont essentiels pour des réactions optimales.
Alors que les hydrogénations catalytiques discontinues traditionnelles utilisent des autoclaves en acier agité ou en acier inoxydable, l’adoption croissante de la technologie des réacteurs à boucle a conduit à une amélioration des performances. Des réacteurs à boucle ont été utilisés avec succès pour l’hydrogénation catalytique de composés nitrés aromatiques par Buss (Bâle, Suisse).
Les avantages par rapport aux autoclaves agités comprennent un transfert de chaleur et de masse amélioré, ainsi qu’une sélectivité de réaction améliorée. L’hydrogénation continue entraîne généralement des durées de cycle discontinues plus courtes et des rendements de produit accrus. De plus, la consommation de catalyseur est souvent réduite.
Les réacteurs à boucle offrent une méthode simple et fiable pour passer de l’usine pilote à la production, car la mise à l’échelle est principalement déterminée par la configuration des buses et le taux de pompage.
En revanche, la mise à l’échelle des autoclaves agités est plus complexe en raison de l’écart important entre les petits navires pilotes et les grands navires de production (par exemple, des rapports de 1:1500 à 1:12500).
Les gradients de température et de concentration, ainsi que les temps de réaction plus longs dans les grands récipients de production, conduisent souvent à des réactions secondaires involontaires et à des rendements de produit réduits.
Les réacteurs à boucle atténuent ces défis en confinant les réactions à une zone bien définie et hautement agitée avec des temps de cycle sensiblement plus courts.
2. Production d’amines aromatiques par substitution nucléophile
Généralement, tous les substituants non carbonés présents sur le cycle aromatique peuvent être remplacés par des groupes amino, mais seules quelques-unes de ces réactions de substitution ont une importance industrielle significative. Ceux-ci inclus:
- Substitution des groupes halogénures
- Substitution des groupes hydroxyle et éther
- Substitution des Sulfo groupes
2.1. Échange d’halogénures
La substitution d’halogénure est utile pour générer des amines contenant des substituants mono-, poly- ou hétérocycliques, ainsi que des diarylamines. De plus, ce procédé s’étend aux arylpolyamines, donnant de nombreux produits pertinents, en particulier en tant qu’intermédiaires pour les colorants et les pigments.
2.1.1. Halogénures activés
Les halogénures aromatiques qui contiennent des groupes attracteurs d’électrons ortho et/ou para, tels que -NO2 ou -CN, peuvent subir des réactions de substitution aromatique nucléophile. L’o-chloronitrobenzène et le p-chloronitrobenzène subissent des réactions avec l’ammoniac à 170 ° C, tandis que le 2,4-dinitrochlorobenzène réagit à une température inférieure de 70 ° C sans nécessiter de catalyseur.
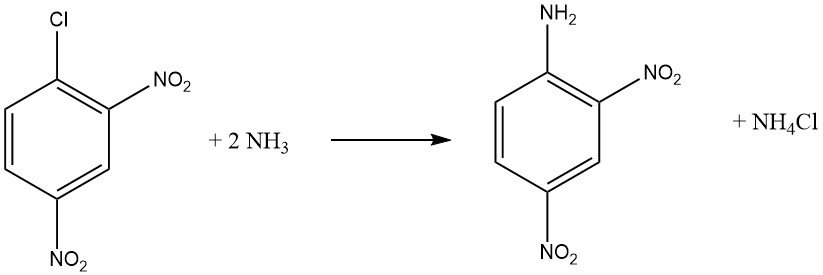
La vitesse de réaction entre le composé haloaromatique et l’amine est influencée par la basicité de l’amine, sauf lorsque l’encombrement stérique est un facteur.
Lors de la réaction de la méthylamine ou de l’ammoniac avec une solution ou une suspension de 2,4-dinitrochlorobenzène à des températures plus élevées, la N-méthyl-2,4-dinitroniline est obtenue avec de bons rendements, au lieu de la 2,4-dinitroaniline.
Conditions de réaction :
La synthèse de la 4-nitroaniline consiste à faire réagir du 4-chloronitrobenzène avec un excès d’ammoniac aqueux dans un réacteur en titane agité à 175 ° C et 4,2 MPa (42 bars) pendant une durée de 10 heures, donnant un rendement de 99,3%.
Un autre exemple est la préparation de 4-(4-méthylanilino)-3-nitrobenzènesulfonamide, obtenue en chauffant du 4-chloro-3-nitrobenzènesulfonamide avec de la p-toluidine à 130 °C pendant 5 heures, ce qui donne un rendement de 90,1 %.
Dans la synthèse de la 4-nitrodiphénylamine, un précurseur des antiozonants du caoutchouc, le 4-nitrochlorobenzène est condensé avec de l’aniline dans du chlorobenzène. Cependant, les dérivés de p-phénylènediamine N-alkylés ne peuvent pas être produits en milieu aqueux.
2.1.2. Halogénures non activés
Les halogénures non activés présentent des réactions d’élimination-addition via un intermédiaire benzyne, un processus exigeant des températures plus élevées que la substitution aromatique nucléophile. De telles réactions donnent des mélanges isomères si des substituants supplémentaires sont présents, limitant leur application industrielle.
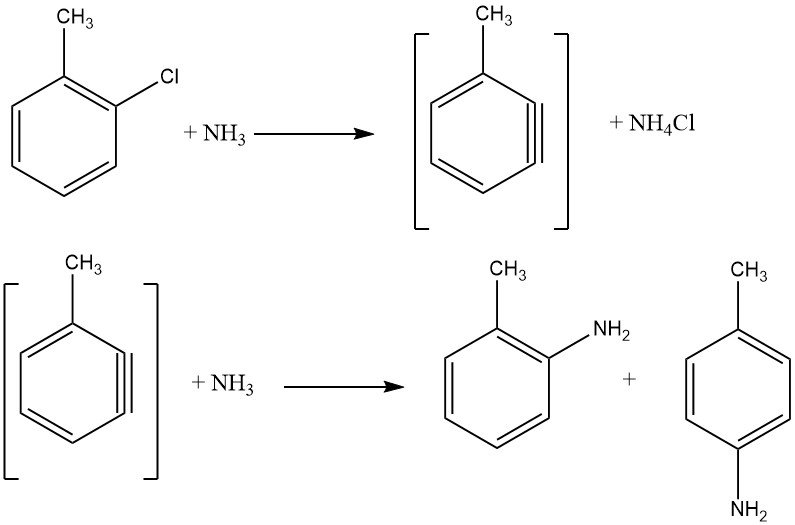
La réaction entre le chlorobenzène et l’ammoniac se produit à des vitesses raisonnables uniquement au-dessus de 200 ° C, nécessitant souvent la présence d’un catalyseur comme les sels de cuivre (I) et de cuivre (II) ou leurs complexes d’amines. Des amides métalliques, tels que NaNH2 et LiNEt2, sont également utilisés. L’énergie nécessaire pour faciliter l’échange d’halogénures est inversement proportionnelle à l’ordre des halogénures activés : F < Cl < Br < I.
Conditions de réaction :
Généralement réalisée dans une suspension ou une solution aqueuse à des températures et pressions élevées, une surveillance attentive de la température est cruciale pendant la phase de chauffage dans les procédés discontinus en raison de la nature exothermique des réactions arylhalogénure-amine.
Les processus continus atténuent généralement cette préoccupation. L’halogénure d’hydrogène libéré est neutralisé en utilisant un excès d’ammoniac ou en ajoutant des bases comme le carbonate de sodium ou l’oxyde de calcium. Avec un excès substantiel d’amine, la réaction se déroule habituellement avec un rendement presque complet.
En cas d’excès insuffisant d’amines fortement basiques dans les solutions aqueuses, des réactions secondaires comme l’hydrolyse ou la formation de diarylamine peuvent se produire.
L’hydrolyse des groupes cyano en amides dans des solutions aqueuses peut être évitée en utilisant des solvants inertes. Les acides carboxyliques peuvent être facilement transformés en amides correspondants en conduisant la réaction dans des milieux non aqueux.
Matériaux de construction :
À des températures et des pressions élevées, la présence d’halogénures et d’amines peut induire une corrosion sévère, ce qui rend difficile la sélection des matériaux et des équipements. Alors que des autoclaves en acier normaux peuvent suffire à des fins de laboratoire, une réduction substantielle de l’épaisseur de paroi est à prévoir (quelques millimètres par an).
De plus, la corrosion sous contrainte peut accélérer la détérioration du réacteur. Les aciers faiblement alliés sont mal adaptés aux unités de production, notamment celles utilisant des réacteurs continus. Des réactions réussies en milieu aqueux ont été obtenues en utilisant des aciers inoxydables, en particulier des aciers austénitiques contenant du chrome et du nickel des types 316.
Cependant, même ces matériaux sont sujets aux piqûres, ce qui conduit à l’utilisation d’alliages plus résistants à la corrosion comme l’Hastelloy et l’Inconel. Les matériaux de réacteur comme le zirconium, le titane et le tantaclad (explosion d’acier recouvert de tantale) ont également trouvé des applications.
2.2. Échange de groupes hydroxyle et éther
Le déplacement d’un groupe hydroxyle aromatique par une amine n’est pas limité aux phénols et aux crésols ; elle s’étend également aux dérivés de la quinoléine et de l’isoquinoléine. L’ammoniac peut remplacer les groupes alcoxy activés en positions ortho ou para, où l’activation est induite par des groupes nitro, halo ou cyano.
Par exemple, le 2,4-dinitroanisole réagit avec l’ammoniac à des températures allant de 50 à 200 °C, donnant de la 2,4-dinitroaniline avec un rendement de 50 à 90 %.
L’intérêt pour les réactions phénol-amine dépend souvent du coût de la matière première phénolique. Les principaux processus d’échange sont :
1. Réaction en milieu aqueux sans catalyseur :
À température et pression élevées, l’acide 3-nitrosalicylique réagit avec l’ammoniac aqueux pour donner de l’acide 2-amino-3-nitrobenzoïque.
2. Réaction en présence d’un catalyseur acide :
Les m-aminophénols se forment par la réaction du résorcinol et de l’hydroxyde d’ammonium aqueux ou des alkylamines, avec l’acide borique comme catalyseur.
En utilisant le même catalyseur dans des conditions anhydres, le résorcinol réagit avec l’éthanolamine ou le 3-amino-1-propanol pour donner des 3-(β- et γ-hydroxyalkylamino)phénols. Un procédé analogue utilise un catalyseur alumine-silice activé par un acide.
3. Les réactions en système multiphase produisent des nitroanilines à partir de nitrophénols et d’ammoniac, de préférence en utilisant un solvant dans lequel le produit de réaction a une bonne solubilité. De nombreux catalyseurs de transfert de phase sont utilisés dans ces réactions.
4. La p-nitrosoaniline et la p-nitroso-N-phénylamine sont synthétisées dans de l’ammoniac liquide en utilisant soit un sel d’ammonium, soit une amine tertiaire dans un solvant organique.
5. Les p-nitrosophénols subissent des réactions avec les alkylamines via les p-nitrosophényléthers, donnant les p-nitrosoanilines N-substituées correspondantes. L’eau produite dans le processus est éliminée pour mener la réaction à son terme.
6. Le procédé Halcon est une importante amination en phase vapeur du phénol impliquant la réaction de phénols ou de naphtols avec de l’ammoniac en phase gazeuse à 250 °C à l’aide d’oxydes métalliques ou non métalliques, tels que MgO, B2O3, Al2O3, SiO2, TiO2 , ou des mélanges.

U.S. Steel Chemicals utilise le procédé Halcon depuis 1982 dans son usine de Haverhill, Ohio, produisant de l’aniline à partir de phénol (capacité d’aniline : 100 000 t/a).
7. La réaction de Bucherer est la réaction de 1- ou 2-naphtols avec des solutions aqueuses d’amine ou d’ammonium en utilisant du sulfite d’hydrogène comme catalyseur. Cette réaction est principalement utilisée pour la série des naphtalènes. L’utilisation de sulfite d’hydrogène réduit la température de réaction de 50 à 150 °C et donne une pureté et un rendement élevés (85 à 95 %).
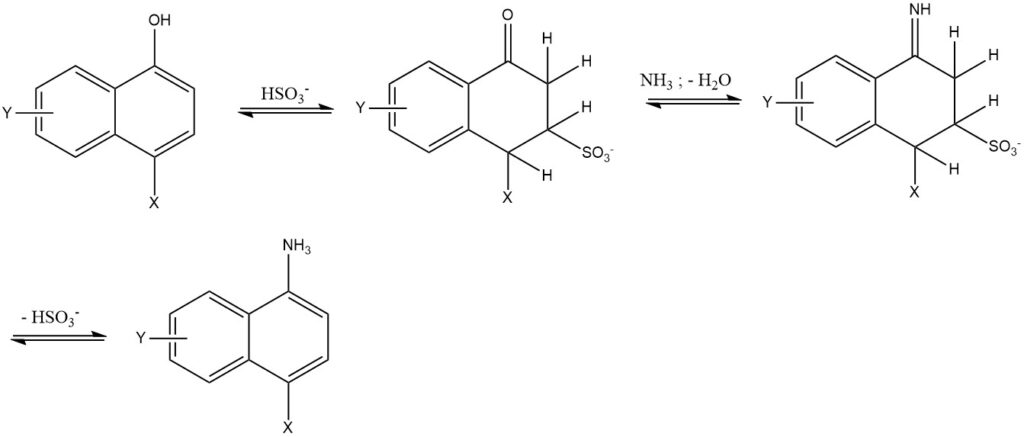
2.3. Échange de groupes Sulfo
L’ammoniac a la capacité de remplacer les groupes sulfo dans les dérivés du benzène, du naphtalène et de l’anthraquinone. Cependant, ce processus d’échange n’a une importance industrielle significative que dans la série des anthraquinones.
Dans le passé, la production de 1-aminoanthraquinones impliquait la réaction de 1-sulfoanthraquinones avec des amines. Pour contrecarrer le dégagement d’hydrogénosulfite, des quantités stoechiométriques d’un agent oxydant, tel que le 3-nitrobenzènesulfonate de sodium, ont été introduites.
Une approche plus récente pour synthétiser la 1-aminoanthraquinone implique la réduction de la nitroanthraquinone correspondante. Cette méthodologie élimine le besoin de la sulfonation catalysée par le mercure conduisant à la 1-sulfoanthraquinone.
Par un procédé conduit en milieu aqueux sous pression à des températures allant de 100 à 200 °C, l’acide 1-cyclohexylaminoanthraquinone-5-sulfonique est généré à partir d’acide anthraquinone-1,5-disulfonique et de cyclohexylamine. L’oxydation du sulfite d’hydrogène est réalisée à l’aide de substances telles que le perchlorate de sodium ou l’acide 3-nitrobenzènesulfonique.
3. Autres processus
Diverses méthodes décrites dans la littérature pour la synthèse des amines aromatiques seront mises en évidence dans cette discussion. Il est important de noter que seule une sélection de ces méthodes sera couverte ici.
La production à grande échelle de 4,4′-méthylènedianiline implique la condensation d’aniline avec du formaldéhyde dans des solutions aqueuses ou aqueuses-méthanoliques.
La diphénylamine est synthétisée par condensation en phase vapeur d’aniline sur un catalyseur d’alumine ou de titane à des températures allant de 450 à 500 °C. Alternativement, il peut être produit en phase liquide à des températures de 175 à 450 °C.
Les arylhydroxylamines peuvent être réarrangées dans de l’acide sulfurique dilué pour former des o- et p-aminophénols.
Un pigment intermédiaire important, la 3,3′-dichlorobenzidine, est fabriqué par Bofors (situé à Muskegan, MI). Le processus implique le réarrangement de la benzidine initié à partir de l’o-chloronitrobenzène.
Référence
- Amines, Aromatic; Ullmann’s Encyclopedia of Industrial Chemistry. – https://onlinelibrary.wiley.com/doi/10.1002/14356007.a02_037
-
Process for the production of aromatic amines. – https://patents.google.com/patent/US5877350A/en