
Producción de aminas aromáticas
Las aminas aromáticas se producen mediante tres tipos de reacciones:
- Reducciones: utilizando elementos metálicos como Hierro (Fe), Zinc (Zn), Estaño (Sn), Aluminio (Al), o sus correspondientes sales; compuestos que contienen azufre; procedimientos electroquímicos; y hidrogenación catalítica.
- Sustituciones nucleofílicas: implican el intercambio de sustituyentes como grupos halógeno, hidroxilo, alcoxi y sulfónico.
- Reordenamientos y degradaciones: incluidas transformaciones como los reordenamientos de bencidina y Beckmann, junto con las degradaciones de Schmidt y Hofmann.
Cabe señalar que los dos primeros tipos de reacciones son más importantes. Los reordenamientos y degradaciones químicas rara vez dan como resultado productos de reacción puros con altos rendimientos.
Tabla de contenido
1. Producción de aminas aromáticas mediante reducción de compuestos nitro
Las aminas aromáticas se producen mediante la reducción de compuestos aromáticos de carbono y nitrógeno que contienen estados de oxidación de nitrógeno que varían de +3 a +2 lata. En particular, la reducción de compuestos nitro se ha utilizado ampliamente a escala industrial debido a la preparación precisa de materiales de partida que se pueden lograr en una amplia gama de compuestos.
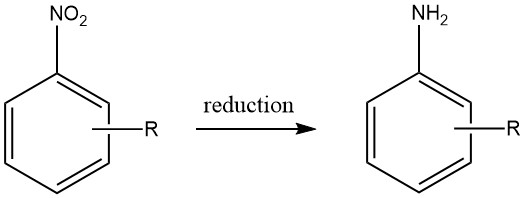
Sin embargo, la viabilidad económica de producir otros compuestos nitrogenados aromáticos como materiales de partida viables para aminas aromáticas es limitada, restringiendo así su utilidad práctica.
1.1. Reducción química
Durante el proceso de reducción química que convierte los compuestos nitro aromáticos en aminas aromáticas, el átomo de hidrógeno que se une al átomo de nitrógeno generalmente se origina en el solvente, a menudo agua, o se introduce mediante el uso de ácido agregado.
Entre los agentes fundamentales para la reducción, tienen prioridad varios metales como el hierro, el estaño y el zinc, junto con el fósforo, los sulfuros, los sulfitos y el dióxido de azufre, que también se utilizan.
Es pertinente señalar que la posible oxidación del agente reductor, que conduce a la formación de un producto de desecho potencialmente inutilizable, requiere medidas cuidadosas de eliminación ambientalmente racionales.
Como resultado, la importancia de la reducción química ha disminuido en comparación con la reducción catalítica, particularmente en la síntesis a gran escala de diversos productos.
1.1.1. Reducción con Hierro
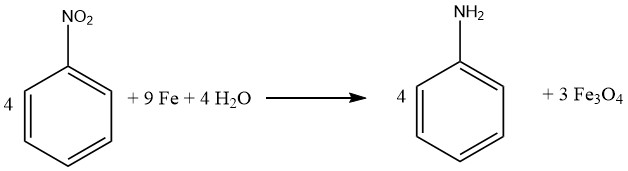
El proceso de reducción de compuestos con hierro, también llamado reducción BÉCHAMP, se dilucidó inicialmente en 1854 y a menudo se lleva a cabo en condiciones ácidas diluidas. El hierro conserva su papel preeminente como catalizador metálico primario para convertir compuestos nitro en sus correspondientes aminas aromáticas.
Prácticamente todos los compuestos nitro se pueden reducir con éxito a aminas aromáticas utilizando hierro en agua y en un medio ácido. En particular, la aparición de subproductos no deseados se anticipa principalmente cuando la molécula contiene otros sustituyentes susceptibles de reducción, como nitroso, azo, hidracina, sulfóxido o grupos nitro adicionales, o si puede sufrir saponificación.
Los enlaces múltiples carbono-carbono permanecen inertes al ataque. Cabe destacar que tanto Bayer (Alemania) como Mobay (Estados Unidos) implementan el enfoque de reducción mediada por hierro para generar óxidos de hierro, produciendo anilina como subproducto.
Otro caso de importancia industrial es el empleo exclusivo de hierro en un medio neutro para reducir el ácido 4,4′-dinitro-2,2′-estilbenodisulfónico para producir ácido 4,4′-diamino-2,2′-estilbenodisulfónico. Además, numerosos tintes intermedios de volumen limitado se reducen mediante el método mediado por hierro.
1.1.2. Reducción con otros metales
Los grupos nitro aromáticos se reducen empleando zinc, estaño y aluminio, o sus sales correspondientes, en medio ácido, neutro o alcalino.
Estos agentes reductores son suaves y, por lo tanto, no interfieren con grupos funcionales como -OH, -OR, -COOH, -CO-Ar, halógeno o -CN. Si bien encuentran aplicaciones en contextos de laboratorio, su utilización práctica carece de importancia industrial sustancial.
Esto se atribuye principalmente al empleo predominante de hidrogenación catalítica, que se alinea con las pautas regulatorias relativas a la gestión de aguas residuales y las prácticas de eliminación.
1.1.3. Reducciones con Sulfuro, Hidrogenosulfito y Dióxido de Azufre
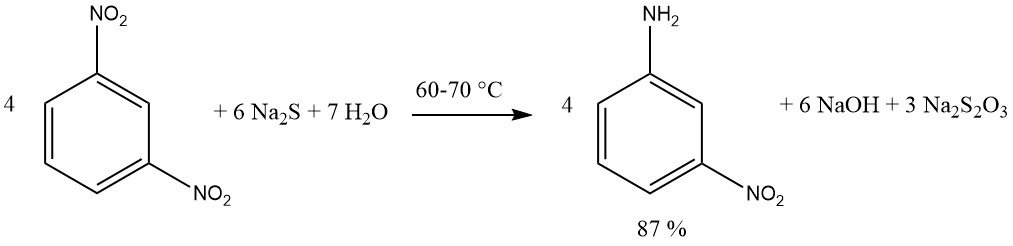
Los compuestos que contienen múltiples grupos nitro se pueden reducir, produciendo nitroaminas, utilizando una cantidad equimolar de reactivo de sulfuro. Se prefieren el sulfuro de sodio o de amonio. Se logra un rendimiento del 87% de m-nitroanilina reduciendo 1,3-dinitrobenceno con sulfuro de sodio.
La adición de azufre elemental disminuye la cantidad necesaria de sulfuro de sodio. Por ejemplo, el 4-nitrotolueno se reduce con alta eficacia para producir 4-aminobenzaldehído utilizando sulfuro de sodio y azufre.
Los grupos azo permanecen inalterados cuando la reducción empleando sulfuro de sodio ocurre dentro del rango de temperatura de 40 a 70 °C, con duraciones de reacción cortas y en ausencia de exceso de sulfuro.
Sin embargo, los sustituyentes halógenos se sustituyen fácilmente por grupos -SH. En los casos en los que un exceso de agente reductor no sea un problema, se puede utilizar hidrogenosulfuro de amonio en presencia de amoníaco.
La reducción de compuestos aromáticos nitro, nitroso o azo a sus correspondientes aminas aromáticas utilizando sulfito o hidrogenosulfito se reconoce como reacción de Piria (PIRIA, 1851).
En condiciones acuosas o alcohólicas se generan predominantemente ácidos N- y C-sulfónicos. Mediante tratamiento con ácido mineral se forma una mezcla de aminas aromáticas y ácidos aminobencenosulfónicos.
La presencia de un intermedio de quinona oxima facilita la introducción de hidrogenosulfito en el anillo aromático durante la reducción de orto y paranitroso-fenoles o naftoles.
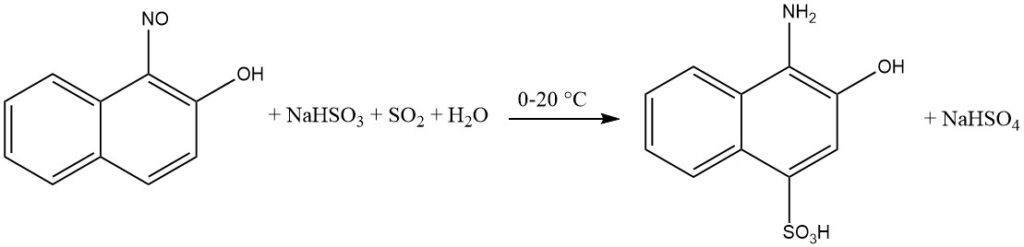
Específicamente, cuando el grupo hidroxilo esté en la posición para, el grupo sulfo se incorporará en la posición 2. Los sustituyentes situados en la posición 3, como Cl o COOH, se sustituyen por hidrógeno.
El uso industrial de la reducción con dióxido de azufre (SO2) se ha vuelto relevante sólo cuando se han reducido las reacciones del lado de la sulfonación de C.
Este procedimiento se lleva a cabo de manera óptima dentro de un sistema sellado, empleando condiciones fuertemente ácidas (por ejemplo, 15 – 40 % de ácido sulfúrico acuoso) y temperaturas que oscilan entre 80 y 180 °C. Los catalizadores como el yodo, el yoduro de hidrógeno o las sales de yodo son integrales y los reactores fabricados con materiales revestidos de vidrio son óptimos.
El ditionito de sodio produce una reducción casi cuantitativa de los grupos aromáticos nitro y nitroso.
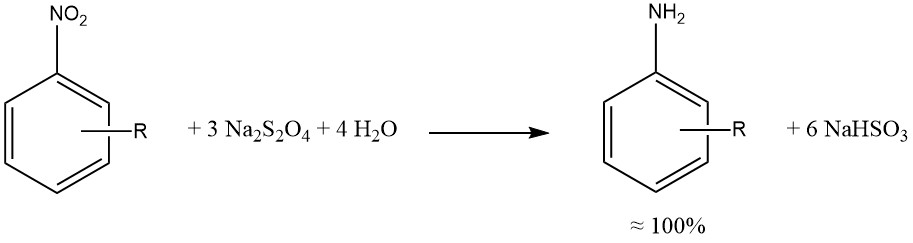
Cuando fue posible, las reducciones por sulfuro, sulfito y otros compuestos de azufre se han reemplazado por hidrogenaciones catalíticas, lo que produce mezclas de reacción más limpias, reducción de la corrosión (asociada con el uso de SO2) y menos preocupaciones ambientales.
1.1.4. Reducción electroquímica de compuestos nitro
La reducción electroquímica suele considerarse una variante especializada de la reducción química. En este enfoque, un compuesto inorgánico sirve como agente reductor y la forma oxidada se reduce posteriormente en el cátodo, permitiéndole reaccionar nuevamente.
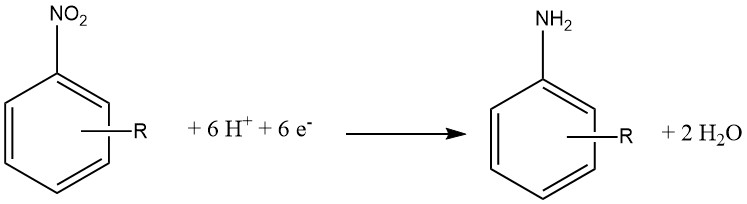
El material del cátodo puede estar hecho de Pb, Sn, Ni o Cu y se utiliza entre un 15 y un 20 % de ácido clorhídrico en el lado del cátodo de una membrana semipermeable. Del lado del ánodo se emplea ácido sulfúrico al 30%.
A pesar de que las reducciones electroquímicas se conocen desde hace más de un siglo, hasta hace poco no se utilizaban ampliamente en aplicaciones comerciales. Sin embargo, el interés en este método está creciendo a medida que continúan mejorando los avances en la tecnología celular y las membranas.
Algunos productos se han producido con éxito mediante reducción electroquímica, particularmente en la India, como el ácido p-aminobenzoico, el p-aminofenol y varios otros, que han alcanzado la escala de planta piloto o más.
1.2. Nitrocompuestos de hidrogenación catalítica
1.2.1. Reducción con hidrazina
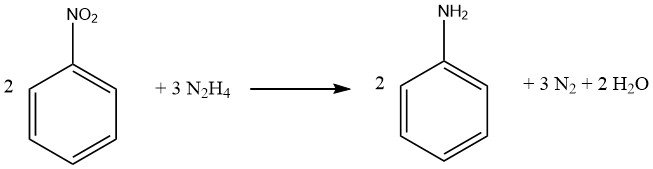
El proceso de reducción de nitrocompuestos aromáticos utilizando hidrazina es una forma especializada de hidrogenación catalítica, en la que la hidrazina sirve como fuente de hidrógeno.
El mecanismo de descomposición de la hidracina en catalizadores de metales preciosos varía según el valor del pH, lo que conduce a un mayor rendimiento de hidrógeno por mol de hidracina a niveles de pH más altos.
En condiciones débilmente alcalinas o neutras se produce 1 mol de H2.
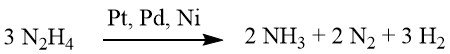
Sin embargo, cuando se complementa con hidróxido de bario o carbonato de calcio, este rendimiento aumenta a 2 moles de H2.
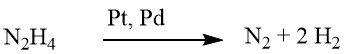
Si bien la reducción con hidracina a veces puede realizarse sin un catalizador, normalmente se lleva a cabo en presencia de un catalizador de hidrogenación, como níquel Raney, paladio sobre carbono o paladio sobre carbonato de calcio.
Idealmente, 1 mol de hidracina proporciona cuatro equivalentes de reducción, dependiendo del pH y el catalizador, aunque prácticamente se generan menos equivalentes, lo que requiere un exceso de hidracina. Es importante destacar que los dobles enlaces carbono-carbono y los grupos carbonilo permanecen inalterados. El aislamiento de intermedios durante el proceso de reducción depende de las condiciones de reacción.
En casos de reducción no catalítica con hidracina, los compuestos que contienen grupos carbonilo se reducen a sus correspondientes compuestos hidroxi mediante la formación de intermedios de hidrazona.
La reducción de compuestos nitro aromáticos usando hidracina, en comparación con la reducción catalítica usando hidrógeno, tiene ventajas limitadas. Encuentra su utilidad principalmente en experimentos de laboratorio y para pequeños volúmenes de productos, donde se pueden emplear condiciones de presión normales.
A escala de laboratorio, el ciclohexeno también se ha explorado como fuente de hidrógeno para la hidrogenación catalítica de compuestos polinitro.
1.2.2. Reducción de Grupos Nitro con Hidrógeno
La generación de mono o poliaminas aromáticas primarias generalmente implica la hidrogenación catalítica del correspondiente nitrocompuesto, ya sea realizada en fase de vapor o en fase líquida, y puede ejecutarse con o sin la presencia de un disolvente. Con este método se fabrican numerosos productos destacados, como anilina, o- y p-toluidina, 2,4- y 2,6-diaminotolueno y 1-naftilamina.
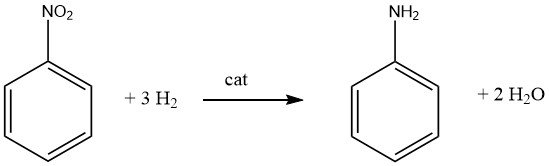
Es importante reconocer que la reducción catalítica de compuestos nitro es exotérmica. Durante la hidrogenación en fase líquida del nitrobenceno, se produce una liberación de 553,5 kJ/mol de energía, mientras que la hidrogenación en fase de vapor a 200 °C libera 493,2 kJ/mol.
Si no se disipa adecuadamente, este calor puede provocar explosiones potencialmente peligrosas, especialmente si se produce una descomposición térmica del compuesto nitro o si se desencadenan reacciones de condensación, como en el caso de los compuestos cloro-nitro.
Para mitigar estos riesgos, se regulan meticulosamente la concentración del compuesto nitro, la cantidad y presión parcial de hidrógeno, la temperatura y la actividad del catalizador.
Hidrogenación en fase de vapor
La aplicación industrial de la hidrogenación en fase de vapor está restringida por el punto de ebullición y la estabilidad térmica de los compuestos nitro.
En los Estados Unidos, una porción sustancial de anilina se produce mediante la hidrogenación en fase de vapor de nitrobenceno utilizando un catalizador de cobre y sílice, lo que produce una impresionante tasa de conversión del 99%. Bayer emplea para ello paladio-alúmina en Europa y Brasil.
Hidrogenación en fase líquida
Generalmente se prefiere la hidrogenación en fase líquida de la mayoría de los compuestos nitro aromáticos. En este escenario, la presión y la temperatura se pueden manipular de forma independiente. Es importante señalar que las temperaturas superiores a 170-200 °C pueden promover reacciones de hidrogenación que involucran al anillo aromático.
Como tal, el proceso de reducción normalmente se lleva a cabo dentro del rango de 100 a 170 °C. Ciertos compuestos, como el 1-bromo-4-nitrobenceno, necesitan temperaturas aún más bajas (20 a 70 °C) para evitar la escisión de grupos sensibles.
Industrialmente se emplean presiones que oscilan entre 1 y 15 MPa (10 y 150 bar). Para la hidrogenación de compuestos nitroaromáticos con mayor sensibilidad, se recomiendan presiones más bajas (0,1–5 MPa, 1–50 bar).
La presión afecta principalmente a la velocidad de reacción al influir en la transferencia de fases y la saturación del catalizador con hidrógeno. El tiempo de reacción depende de varios parámetros, incluida la presión, concentración, temperatura, actividad y concentración del catalizador y mezclado.
A menudo se observa un período de inducción inicial, que no se ve afectado por la actividad del catalizador. Los tiempos de reacción suelen oscilar entre unos pocos minutos y varias horas. Se requieren duraciones más largas si estos parámetros no se optimizan adecuadamente.
Debido a la naturaleza inherentemente exotérmica de la reacción, se deben seguir estrictas precauciones de seguridad, particularmente en la hidrogenación industrial de compuestos polinitro aromáticos en ausencia de disolventes.
El carácter exotérmico se gestiona mediante la adición continua de pequeñas cantidades del compuesto nitro, manteniendo su concentración por debajo del 2%. Se puede introducir agua desionizada para absorber el calor de la reacción mediante evaporación continua.
Esto no sólo mitiga los problemas relacionados con el calor sino que también afecta la actividad del catalizador; un mayor contenido de agua en la hidrogenación discontinua de dinitrotolueno sin disolvente conduce a una menor actividad del catalizador de Pd sobre carbono.
Si la amina es soluble en agua, entonces se puede emplear como disolvente. Este enfoque también es adecuado cuando el compuesto nitro forma sales solubles en agua con álcalis, como se ve con los ácidos nitrocarbónico o sulfónico.
En ciertos casos, las soluciones de la amina en agua (30-40%) se pueden generar directamente dentro del reactor de hidrogenación, evitando la necesidad de pasos de concentración adicionales.
Generalmente se prefieren los disolventes, como el metanol y el 2-propanol. También se han utilizado dioxano, tetrahidrofurano y N-metilpirrolidona. Por ejemplo, el o-nitrofenol logra buenos rendimientos de hidrogenación en metanol utilizando Pt sobre carbono.
Cuando se utiliza un disolvente inmiscible en agua, como el tolueno, se debe minimizar el contenido de agua para preservar la actividad del catalizador, de forma similar a la hidrogenación sin disolvente. En algunos casos, los compuestos nitro altamente polares, como la bis(4-amino-3-nitrofenil)sulfona, se hidrogenan con amoníaco líquido.
Catalizadores de hidrogenación
La hidrogenación en fase de vapor se basa en metales o derivados metálicos sobre soportes dentro de lechos fijos o lechos fluidizados. La hidrogenación en fase líquida emplea predominantemente metales con áreas superficiales extensas, como el níquel Raney, el níquel-hierro Raney, el cobalto Raney y el cobre Raney, debido a su costo relativamente más bajo.
Los catalizadores de metales preciosos, como Pt y Pd, se utilizan generalmente en concentraciones de 0,5 a 5 % en peso sobre materiales de soporte de alta superficie como carbón, sílice, óxido de aluminio o carbonatos alcalinotérreos.
La forma de catalizador en polvo es adecuada para lodos, mientras que la forma de pellets se utiliza en lechos fijos. Es imperativo optimizar las condiciones que permitan la reutilización y regeneración del catalizador, además de que sean económicos para la producción a gran escala (>500 t/a).
Los subproductos de descomposición y oxidación frecuentemente conducen a la desactivación del catalizador por bloqueo de la superficie. Incluso pequeñas cantidades de compuestos de azufre, arsénico o antimonio pueden envenenar los catalizadores. Sin embargo, estos venenos para catalizadores exhiben menos impacto a medida que aumentan los estados de oxidación (por ejemplo, As3- → As5+ o S2- → S4+).
Con catalizadores de metales nobles, los iones de haluro, sodio, magnesio, cobalto y CO2 pueden reducir la actividad. Los compuestos nitroaromáticos que contienen azufre demuestran una mejor reducción usando molibdeno o sulfuro de tungsteno, o con catalizadores de níquel que contienen zinc o carbonato de calcio.
El Pd sobre catalizadores de carbono puede verse afectado por las sales de cobre, mientras que tanto el nitrofenol como el nitrocresol actúan como fuertes venenos para los catalizadores y activadores de la descomposición durante la hidrogenación del dinitrotolueno.
Hidrogenación de compuestos nitro sustituidos con haluros
La hidrogenación de compuestos nitro con sustituciones de haluro a menudo requiere temperaturas más bajas para evitar el intercambio haluro-hidrógeno. Los catalizadores, particularmente los catalizadores metálicos envenenados con sulfuro, exhiben una actividad reducida en tales condiciones.
El 1-cloro-2-nitrobenceno se puede reducir eficazmente a 80 °C y 1,0 a 5,0 MPa (10 a 50 bar) utilizando un catalizador de Pt al 5 % en peso sobre carbono tratado con ácido sulfúrico diluido, seguido de H2 y H2S. Este catalizador se puede reciclar hasta 30 veces sin purificación adicional.
Se han empleado metodologías similares para la hidrogenación de 3-cloronitrobenceno utilizando catalizadores tratados con tiocianato. El uso de catalizadores modificados mejora la selectividad y eficacia de la reacción, lo que da como resultado altos rendimientos y una excelente pureza del producto.
Equipos y Materiales de Construcción
La hidrogenación en fase de vapor generalmente se realiza en modo continuo, mientras que la hidrogenación en fase líquida se puede lograr mediante procesos discontinuos o continuos, según la escala. La hidrogenación continua en fase líquida se emplea principalmente para unos pocos productos seleccionados a gran escala, como dinitrotolueno, nitrotolueno y 1-nitronaftaleno.
Sin embargo, la mayor parte de la producción de aminas aromáticas implica la hidrogenación por lotes utilizando suspensiones de catalizador. Este enfoque permite transiciones rápidas entre productos, un lujo que no se puede permitir cuando se utilizan catalizadores de lecho fijo.
Es importante mezclar eficazmente las tres fases (gas hidrógeno, solución de compuesto nitro y catalizador sólido). El transporte rápido de reactivos a los centros catalíticos activos y la posterior eliminación del producto son vitales para reacciones óptimas.
Si bien las hidrogenaciones catalíticas por lotes tradicionales utilizan autoclaves de acero agitado o de acero inoxidable, la creciente adopción de la tecnología de reactor de bucle ha llevado a un mejor rendimiento. Buss (Basilea, Suiza) ha empleado con éxito reactores de bucle para la hidrogenación catalítica de compuestos nitro aromáticos.
Las ventajas sobre los autoclaves agitados incluyen una mayor transferencia de calor y masa, así como una mejor selectividad de reacción. La hidrogenación continua generalmente da como resultado tiempos de ciclo por lotes más cortos y mayores rendimientos del producto. Además, a menudo se reduce el consumo de catalizador.
Los reactores de circuito ofrecen un método sencillo y confiable para la transición de la planta piloto a la producción, ya que la ampliación está determinada principalmente por la configuración de las boquillas y la tasa de bombeo.
Por el contrario, la ampliación de escala para autoclaves agitados es más compleja debido a la discrepancia significativa entre los recipientes piloto pequeños y los recipientes de producción grandes (por ejemplo, proporciones de 1:1500 a 1:12500).
Los gradientes de temperatura y concentración, junto con tiempos de reacción más prolongados en recipientes de producción más grandes, a menudo provocan reacciones secundarias no deseadas y rendimientos reducidos del producto.
Los reactores de circuito mitigan estos desafíos al limitar las reacciones a una zona bien definida y altamente agitada con tiempos de ciclo sustancialmente más cortos.
2. Producción de aminas aromáticas por sustitución nucleofílica
Generalmente, todos los sustituyentes distintos del carbono presentes en el anillo aromático pueden reemplazarse por grupos amino, pero sólo unas pocas de estas reacciones de sustitución tienen una importancia industrial significativa. Éstas incluyen:
- Sustitución de grupos halogenuros
- Sustitución de grupos hidroxilo y éter.
- Sustitución de grupos sulfo
2.1. Intercambio de halogenuros
La sustitución de haluro es útil para generar aminas que contienen sustituyentes mono, poli o heterocíclicos, junto con diarilaminas. Además, este proceso se extiende a las arilpoliaminas, dando lugar a numerosos productos de relevancia, particularmente como intermedios para tintes y pigmentos.
2.1.1. Haluros activados
Los haluros aromáticos que contienen grupos aceptores de electrones orto y/o para, como -NO2 o -CN, pueden sufrir reacciones de sustitución aromática nucleofílica. Tanto el o-cloronitrobenceno como el p-cloronitrobenceno reaccionan con amoníaco a 170 °C, mientras que el 2,4-dinitrobenceno reacciona a una temperatura más baja de 70 °C sin necesidad de catalizador.
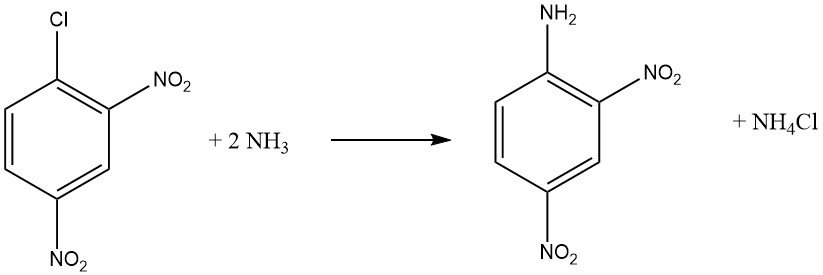
La velocidad de reacción entre el compuesto haloaromático y la amina está influenciada por la basicidad de la amina, excepto cuando el impedimento estérico es un factor.
Al hacer reaccionar metilamina o amoníaco con una solución o suspensión de 2,4-dinitroclorobenceno a temperaturas más altas, se obtiene N-metil-2,4-dinitronilina con buenos rendimientos, en lugar de 2,4-dinitroanilina.
Condiciones de reacción:
La síntesis de 4-nitroanilina implica hacer reaccionar 4-cloronitrobenceno con un exceso de amoníaco acuoso en un reactor de titanio agitado a 175 °C y 4,2 MPa (42 bar) durante 10 horas, con un rendimiento del 99,3%.
Otro ejemplo es la preparación de 4-(4-metilanilino)-3-nitrobencenosulfonamida, que se logra calentando 4-cloro-3-nitrobencenosulfonamida con p-toluidina a 130 °C durante 5 horas, lo que da como resultado un rendimiento del 90,1 %.
En la síntesis de 4-nitrodifenilamina, un precursor de los antiozonantes del caucho, el 4-nitroclorobenceno se condensa con anilina en clorobenceno. Sin embargo, los derivados de p-fenilendiamina N-alquilados no pueden producirse en medio acuoso.
2.1.2. Haluros no activados
Los haluros inactivados exhiben reacciones de eliminación-adición a través de un intermediario bencino, un proceso que exige temperaturas más altas que la sustitución aromática nucleofílica. Estas reacciones producen mezclas isoméricas si hay sustituyentes adicionales presentes, lo que limita su aplicación industrial.
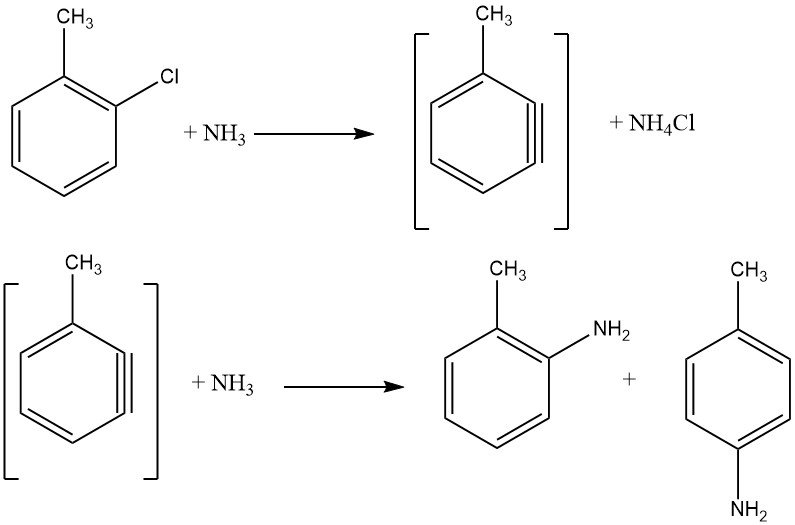
La reacción entre el clorobenceno y el amoníaco se produce a velocidades razonables sólo por encima de 200 °C, y a menudo requiere la presencia de un catalizador como las sales de cobre (I) y cobre (II) o sus complejos de amina. También se emplean amidas metálicas como NaNH2 y LiNEt2. La energía necesaria para facilitar el intercambio de haluros es inversamente proporcional al orden de los haluros activados: F < Cl < Br < I.
Condiciones de reacción:
Generalmente realizado en una suspensión o solución acuosa a temperaturas y presiones elevadas, el control cuidadoso de la temperatura es crucial durante la fase de calentamiento en procesos discontinuos debido a la naturaleza exotérmica de las reacciones de haluro de ariluro-amina.
Los procesos continuos generalmente mitigan esta preocupación. El haluro de hidrógeno liberado se neutraliza utilizando un exceso de amoníaco o añadiendo bases como carbonato de sodio u óxido de calcio. Con un exceso sustancial de amina, la reacción normalmente transcurre con un rendimiento casi completo.
En caso de un exceso insuficiente de aminas fuertemente básicas en soluciones acuosas, pueden ocurrir reacciones secundarias como hidrólisis o formación de diarilamina.
La hidrólisis de grupos ciano a amidas en soluciones acuosas se puede evitar utilizando disolventes inertes. Los ácidos carboxílicos se pueden transformar fácilmente en las amidas correspondientes realizando la reacción en medios no acuosos.
Materiales de construcción:
A temperaturas y presiones elevadas, la presencia de haluros y aminas puede inducir una corrosión severa, lo que hace que la selección de materiales y equipos sea un desafío. Si bien los autoclaves de acero normales pueden ser suficientes para fines de laboratorio, se puede esperar una reducción sustancial del espesor de la pared (unos pocos milímetros por año).
Además, la corrosión bajo tensión puede acelerar el deterioro del reactor. Los aceros de baja aleación no son adecuados para las unidades de producción, en particular aquellas que emplean reactores continuos. Se han logrado reacciones exitosas en medios acuosos utilizando aceros inoxidables, específicamente aceros austeníticos que contienen cromo y níquel del tipo 316.
Sin embargo, incluso estos materiales son propensos a sufrir picaduras, lo que lleva a la utilización de aleaciones más resistentes a la corrosión como Hastelloy e Inconel. Los materiales de reactores como el circonio, el titanio y el tantalio (acero revestido por explosión con tantalio) también han encontrado aplicaciones.
2.2. Intercambio de grupos hidroxilo y éter
El desplazamiento de un grupo hidroxilo aromático por una amina no se limita a los fenoles y cresoles; se extiende también a los derivados de quinolina e isoquinolina. El amoníaco puede sustituir grupos alcoxi activados en posiciones orto o para, donde la activación es inducida por grupos nitro, halo o ciano.
Por ejemplo, el 2,4-dinitroanisol reacciona con amoníaco a temperaturas que oscilan entre 50 y 200 °C, produciendo 2,4-dinitroanilina con un rendimiento del 50 al 90%.
El interés en las reacciones fenol-amina depende a menudo del coste de la materia prima del fenol. Los principales procesos de intercambio son:
1. Reacción en medio acuoso sin catalizador:
A temperatura y presión elevadas, el ácido 3-nitrosalicílico reacciona con amoníaco acuoso para producir ácido 2-amino-3-nitrobenzoico.
2. Reacción en presencia de un catalizador ácido:
Los m-aminofenoles se forman mediante la reacción de resorcinol e hidróxido de amonio acuoso o alquilaminas, con ácido bórico como catalizador.
Al emplear el mismo catalizador en condiciones anhidras, el resorcinol reacciona con etanolamina o 3-amino-1-propanol para producir 3-(β- y γ-hidroxialquilamino)fenoles. Un proceso análogo utiliza un catalizador de sílice-alúmina activado con ácido.
3. Las reacciones de sistemas multifásicos producen nitroanilinas a partir de nitrofenoles y amoníaco, preferiblemente empleando un disolvente en el que el producto de reacción tenga buena solubilidad. En estas reacciones se emplean numerosos catalizadores de transferencia de fases.
4. La p-nitrosoanilina y la p-nitroso-N-fenilamina se sintetizan en amoníaco líquido utilizando una sal de amonio o una amina terciaria en un disolvente orgánico.
5. Los p-nitrosofenoles sufren reacciones con alquilaminas a través de éteres de p-nitrosofenilo, produciendo las correspondientes p-nitrosoanilinas N-sustituidas. El agua producida en el proceso se elimina para completar la reacción.
6. El proceso Halcon es una importante aminación de fenol en fase de vapor que implica la reacción de fenoles o naftoles con amoníaco en fase gaseosa a 250 °C utilizando óxidos metálicos o no metálicos, como MgO, B2O3, Al2O3, SiO2, TiO2. , o mezclas.

U.S. Steel Chemicals ha empleado el proceso Halcon desde 1982 en su planta de Haverhill, Ohio, produciendo anilina a partir de fenol (capacidad de anilina: 100.000 t/a).
7. La reacción de Bucherer es la reacción de 1 o 2 naftoles con soluciones acuosas de amina o amonio utilizando sulfito de hidrógeno como catalizador. Esta reacción se utiliza principalmente para la serie de naftaleno. El uso de sulfito de hidrógeno reduce la temperatura de reacción entre 50 y 150 °C y produce una alta pureza y rendimiento (85 – 95%).
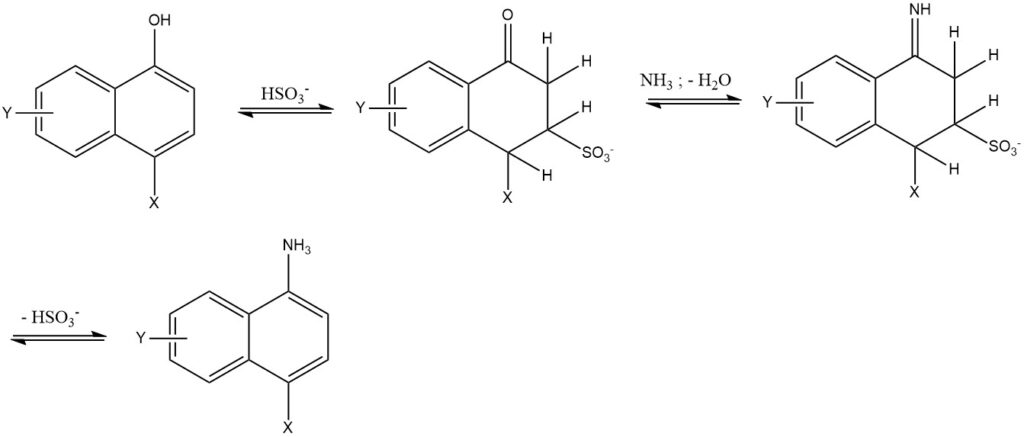
2.3. Intercambio de Grupos Sulfo
El amoníaco tiene la capacidad de sustituir grupos sulfo en derivados de benceno, naftaleno y antraquinona. Sin embargo, este proceso de intercambio tiene una importancia industrial significativa sólo dentro de la serie de las antraquinonas.
En el pasado, la producción de 1-aminoantraquinonas implicaba la reacción de 1-sulfoantraquinonas con aminas. Para contrarrestar la liberación de sulfito de hidrógeno se introdujeron cantidades estequiométricas de un agente oxidante, como por ejemplo 3-nitrobencenosulfonato de sodio.
Un enfoque más reciente para sintetizar 1-aminoantraquinona implica la reducción de la nitroantraquinona correspondiente. Esta metodología elimina la necesidad de la sulfonación catalizada por mercurio que conduce a 1-sulfoantraquinona.
Mediante un proceso realizado en un medio acuoso bajo presión a temperaturas que oscilan entre 100 y 200 °C, se genera ácido 1-ciclohexilaminoantraquinona-5-sulfónico a partir del ácido antraquinona-1,5-disulfónico y la ciclohexilamina. La oxidación del sulfito de hidrógeno se logra utilizando sustancias como el perclorato de sodio o el ácido 3-nitrobencenosulfónico.
3. Otros procesos
En esta discusión se destacarán varios métodos descritos en la literatura para la síntesis de aminas aromáticas. Es importante tener en cuenta que aquí sólo se cubrirá una selección de estos métodos.
La producción a gran escala de 4,4′-metilendianilina implica la condensación de anilina con formaldehído en soluciones acuosas o metanólicas acuosas.
La difenilamina se sintetiza mediante condensación en fase de vapor de anilina sobre un catalizador de alúmina o titanio a temperaturas que oscilan entre 450 y 500 °C. Alternativamente, se puede producir en fase líquida a temperaturas de 175 a 450 °C.
Las arilhidroxilaminas se pueden reordenar en ácido sulfúrico diluido para formar o- y p-aminofenoles.
Un pigmento intermedio importante, la 3,3′-diclorobencidina, es fabricado por Bofors (ubicada en Muskegan, MI). El proceso implica la transposición de bencidina iniciada a partir de o-cloronitrobenceno.
Referencias
- Amines, Aromatic; Ullmann’s Encyclopedia of Industrial Chemistry. – https://onlinelibrary.wiley.com/doi/10.1002/14356007.a02_037
-
Process for the production of aromatic amines. – https://patents.google.com/patent/US5877350A/en