
Production of Aromatic Amines
Aromatic amines are produced by three types of reactions:
- Reductions: using metallic elements like Iron (Fe), Zinc (Zn), Tin (Sn), Aluminum (Al), or their corresponding salts; sulfur-containing compounds; electrochemical procedures; and catalytic hydrogenation.
- Nucleophilic substitutions: involving the exchange of substituents like halogen, hydroxyl, alkoxy, and sulfonic groups.
- Rearrangements and degradations: including transformations such as the benzidine and Beckmann rearrangements, along with the Schmidt and Hofmann degradations.
It should be noted that the first two reaction types are more important. Chemical rearrangements and degradations rarely result in pure reaction products with high yields.
Table of Contents
1. Production of Aromatic amines by Reduction of Nitro Compounds
Aromatic amines are produced by reduction of aromatic carbon-nitrogen compounds containing nitrogen oxidation states ranging from +3 to +2 can. Notably, the reduction of nitro compounds has been widely used on an industrial scale due to the precise preparation of starting materials that can be achieved across a diverse range of compounds.
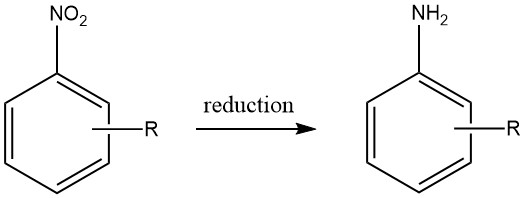
However, the economic feasibility of producing other aromatic nitrogen compounds as viable starting materials for aromatic amines is limited, thereby restricting their practical utility.
1.1. Chemical Reduction
During the chemical reduction process that converts aromatic nitro compounds into aromatic amines, the hydrogen atom that becomes bonded to the nitrogen atom typically originates from the solvent, often water, or is introduced by the use of added acid.
Among the pivotal agents for reduction, various metals like iron, tin, and zinc take precedence, along with phosphorus, sulfides, sulfites, and sulfur dioxide, which also are used.
It is pertinent to note that the potential oxidation of the reducing agent, leading to the formation of a potentially unusable waste product, necessitates careful environmentally sound disposal measures.
As a result, the significance of chemical reduction has diminished in comparison to catalytic reduction, particularly in the large-scale synthesis of various products.
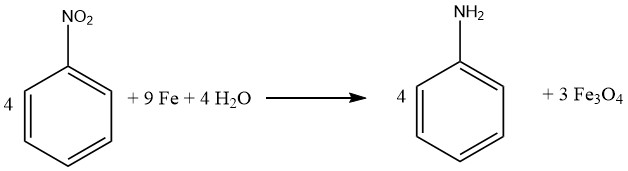
1.1.1. Reduction with Iron
The process of reducing compounds with iron also called BÉCHAMP reduction was initially elucidated in 1854 and is often carried out in dilute acidic conditions, . Iron retains its preeminent role as the primary metal catalyst for converting nitro compounds into their corresponding aromatic amines.
Virtually all nitro compounds can be successfully reduced to aromatic amines using iron in water and acid milieu. Notably, the emergence of undesired byproducts is primarily anticipated when the molecule contains other substituents susceptible to reduction, such as nitroso, azo, hydrazine, sulfoxide, or additional nitro groups, or if it can undergo saponification.
Carbon-carbon multiple bonds remain inert to attack. Prominently, both Bayer (Germany) and Mobay (United States) implement the iron-mediated reduction approach for generating iron oxides, yielding aniline as a byproduct.
Another case of industrial significance is the exclusive employment of iron in a neutral milieu for reducing 4,4′-dinitro-2,2′-stilbenedisulfonic acid to yield 4,4′-diamino-2,2′-stilbenedisulfonic acid. Also, numerous dye intermediates of limited volume undergo reduction via the iron-mediated method.
1.1.2. Reduction with Other Metals
Aromatic nitro groups undergo reduction by employing zinc, tin, and aluminum, or their corresponding salts, in acidic, neutral, or alkaline medium.
These reductive agents are mild and thereby do not interfere with functional groups such as -OH, -OR, -COOH, -CO-Ar, halogen, or -CN. While they find applications in laboratory contexts, their practical utilization lacks substantial industrial significance.
This is primarily attributed to the prevalent employment of catalytic hydrogenation, which aligns with regulatory guidelines concerning wastewater management and disposal practices.
1.1.3. Reductions with Sulfide, Hydrogensulfite and Sulfur Dioxide
Compounds containing multiple nitro groups can be reduced, yielding nitro amines, by using an equimolar quantity of sulfide reagent. Sodium or ammonium sulfide are preferred. A yield of 87% m-nitroaniline is achieved by reducing 1,3-dinitrobenzene with sodium sulfide.
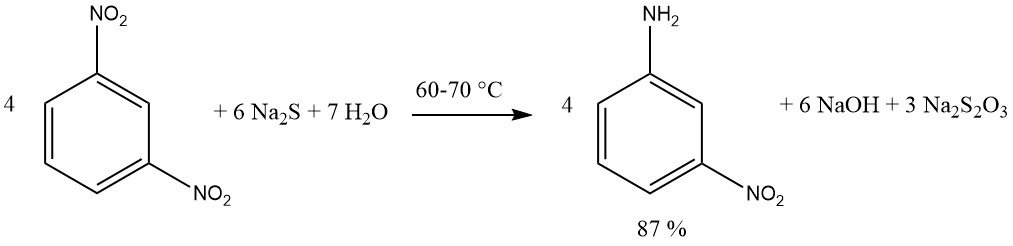
The addition of elemental sulfur diminishes the requisite amount of sodium sulfide. For exemple, 4-Nitrotoluene is reduced with high efficiency to yield 4-aminobenzaldehyde using sodium sulfide and sulfur.
Azo groups remain unaltered when reduction employing sodium sulfide occurs within the temperature range of 40 – 70 °C, with short reaction durations, and in the absence of sulfide excess.
However, halogen substituents are readily substituted by -SH groups. In cases where an excess of the reducing agent is not a problem, ammonium hydrogensulfide in the presonce of ammonia can be used.
The reduction of aromatic nitro, nitroso, or azo compounds to their corresponding aromatic amines using sulfite or hydrogensulfite is recognized as the Piria reaction (PIRIA, 1851).
Under aqueous or alcoholic conditions, predominantly N- and C-sulfonic acids are generated. By treatment with mineral acid, a mixture of aromatic amines and aminobenzenesulfonic acids is formed.
The presence of a quinone oxime intermediate facilitates the introduction of hydrogensulfite onto the aromatic ring during the reduction of ortho- and para-nitroso-phenols or naphthols.
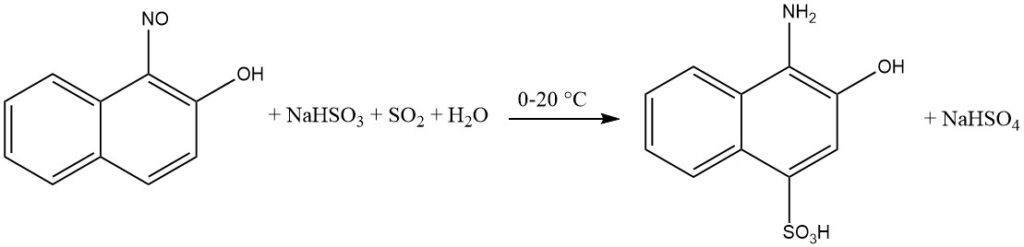
Specifically, when the hydroxyl group is in the para position, the sulfo group will be incorporated in the 2-position. Substituents positioned at the 3-position, such as Cl or COOH, are replaced by hydrogen.
The industrial use of reduction with sulfur dioxide (SO2) has became relevant only when the C-sulfonation side reacation have been reduced .
This procedure is optimally conducted within a sealed system, employing strongly acidic conditions (e.g., 15 – 40% aqueous sulfuric acid), and temperatures spanning 80 – 180 °C. Catalysts such as iodine, hydrogen iodide, or iodine salts are integral, and reactors fabricated from glass-lined materials are optimal.
Sodium dithionite yiels nearly quantitative reduction of the aromatic nitro and nitroso groups.
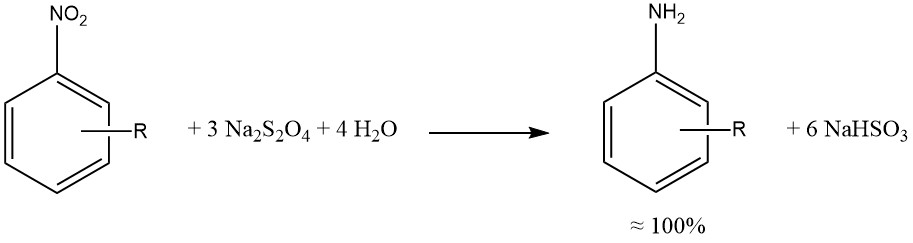
When possible, reductions by sulfide, sulfite, and other sulfur compounds have been replaced by catalytic hydrogenations, yielding cleaner reaction mixtures, reduced corrosion (associated with SO2 usage), and fewer environmental concerns.
1.1.4. Electrochemical Reduction of Nitro Compounds
Electrochemical reduction is typically considered a specialized variant of chemical reduction. In this approach, an inorganic compound serves as the reducing agent, and the oxidized form is subsequently reduced at the cathode, enabling it to react again.
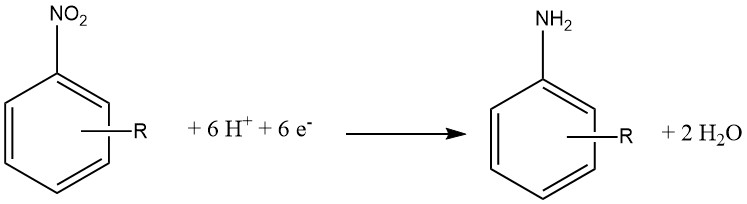
The cathode material can be made of Pb, Sn, Ni, or Cu and 15 – 20% hydrochloric acid is used on the cathode side of a semi-permeable membrane. On the anode side, 30% sulfuric acid is employed.
Despite electrochemical reductions being known for over a century, they were not widely employed in commercial applications until recently. However, the interest in this method is growing as advancements in cell technology and membranes continue to improve.
Some products have been successfully produced by electrochemical reduction, particularly in India, such as p-aminobenzoic acid, p-aminophenol, and several others, which have reached pilot-plant scale or beyond.
1.2. Catalytic Hydrogenation Nitro Compounds
1.2.1. Reduction with Hydrazine
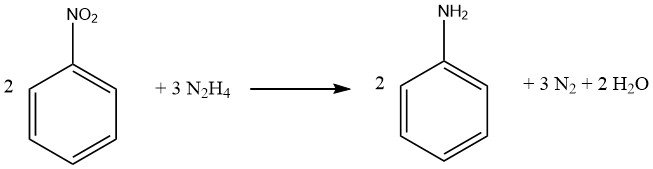
The reduction process of aromatic nitro compounds using hydrazine is a specialized form of catalytic hydrogenation, wherein hydrazine serves as the hydrogen source.
The decomposition mechanism of hydrazine on precious-metal catalysts varies based on the pH value, leading to an increased hydrogen yield per mole of hydrazine at higher pH levels.
Under weakly alkaline or neutral conditions, 1 mole of H2 is produced.
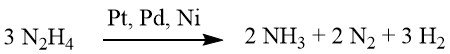
However, when supplemented with barium hydroxide or calcium carbonate, this yield escalates to 2 moles of H2.
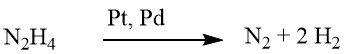
While reduction using hydrazine can sometimes proceed without a catalyst, it is typically conducted in the presence of a hydrogenation catalyst, such as Raney nickel, palladium on carbon, or palladium on calcium carbonate.
Ideally 1 mole of hydrazine provides four reduction equivalents, contingent upon pH and catalyst, though fewer equivalents are practically generated, necessitating an excess of hydrazine. Importantly, carbon-carbon double bonds and carbonyl groups remain unaltered. The isolation of intermediates during the reduction process depends on reaction conditions.
In cases of non-catalytic reduction with hydrazine, compounds containing carbonyl groups are reduced to their corresponding hydroxy compounds via the formation of hydrazone intermediates.
The reduction of aromatic nitro compounds using hydrazine, in comparison to catalytic reduction employing hydrogen, holds limited advantages. It finds its utility primarily in laboratory experiments and for small product volumes, where normal pressure conditions can be employed.
On a laboratory scale, cyclohexene has also been explored as a hydrogen source for the catalytic hydrogenation of polynitro compounds.
1.2.2. Reduction of Nitro Groups with Hydrogen
The generation of primary aromatic mono- or polyamines generally entails catalytic hydrogenation of the corresponding nitro compound, either conducted in the vapor phase or liquid phase, and can be executed with or without the presence of a solvent. Numerous prominent products, including aniline, o- and p-toluidine, 2,4- and 2,6-diaminotoluene, and 1-naphthylamine, are manufactured using this method.
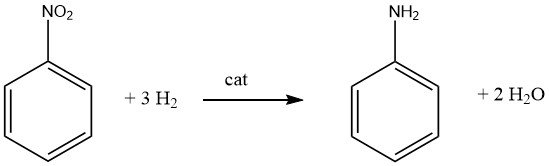
It’s important to recognize that catalytic reduction of nitro compounds is exothermic. During the liquid-phase hydrogenation of nitrobenzene, a release of 553.5 kJ/mol of energy occurs, while vapor-phase hydrogenation at 200 °C liberates 493.2 kJ/mol.
If not adequately dissipated, this heat can lead to potentially hazardous explosions, particularly if thermal decomposition of the nitro compound transpires or if condensation reactions are triggered, as in the case of chloro-nitro compounds.
To mitigate these risks, the concentration of the nitro compound, the quantity and partial pressure of hydrogen, the temperature, and the catalyst’s activity are meticulously regulated.
Vapor-Phase Hydrogenation
The industrial application of vapor-phase hydrogenation is restricted by the boiling point and thermal stability of nitro compounds.
In the United States, a substantial portion of aniline is produced via vapor-phase hydrogenation of nitrobenzene using a copper–silica catalyst, yielding an impressive 99% conversion rate. Bayer employs Pd–alumina for this purpose in Europe and Brazil.
Liquid-Phase Hydrogenation
The liquid-phase hydrogenation of most aromatic nitro compounds is generally preferred. In this scenario, pressure and temperature can be independently manipulated. It’s important to note that temperatures exceeding 170–200 °C can promote hydrogenation reactions involving the aromatic ring.
As such, the reduction process is typically carried out within the range of 100–170 °C. Certain compounds, such as 1-bromo-4-nitrobenzene, necessitate even lower temperatures (20–70 °C) to prevent sensitive group cleavage.
Industrially, pressures ranging from 1–15 MPa (10–150 bar) are employed. For the hydrogenation of nitro aromatic compounds with heightened sensitivity, lower pressures (0.1–5 MPa, 1–50 bar) are recommended.
Pressure primarily affects the reaction rate by influencing phase transfer and the saturation of the catalyst with hydrogen. Reaction time hinges upon various parameters, including hydrogen pressure, concentration, temperature, catalyst activity and concentration, and mixing.
Often, an initial induction period is observed, which remains unaffected by catalyst activity. Reaction times typically span from a few minutes to several hours. Longer durations are required if these parameters are not suitably optimized.
Due to the inherently exothermic nature of the reaction, stringent safety precautions must be adhered to, particularly in the industrial hydrogenation of aromatic polynitro compounds in the absence of solvents.
The exothermic nature is managed by the continuous addition of small quantities of the nitro compound, maintaining its concentration below 2%. Deionized water can be introduced to absorb reaction heat through continuous evaporation.
This not only mitigates heat-related issues but also affects catalyst activity; higher water content in the batch hydrogenation of dinitrotoluene without a solvent leads to decreased activity of the Pd on carbon catalyst.
If the amine is soluble in water then it can be employed as the solvent. This approach is also suitable when the nitro compound forms water-soluble salts with alkali, as seen with nitrocarbonic or sulfonic acids.
In certain cases, solutions of the amine in water (30–40%) can be directly generated within the hydrogenation reactor, obviating the need for additional concentration steps.
Solvents, such as methanol and 2-propanol, are typically favored. Dioxane, tetrahydrofuran, and N-methylpyrrolidone have also been utilized. For instance, o-nitrophenol achieves good hydrogenation yields in methanol utilizing Pt on carbon.
When using a water-immiscible solvent, such as toluene, the water content must be minimized to preserve catalyst activity, similar to solvent-free hydrogenation. In some cases, highly polar nitro compounds, like bis(4-amino-3-nitrophenyl)sulfone, are hydrogenated using liquid ammonia.
Hydrogenation Catalysts
Vapor-phase hydrogenation relies on metals or metal derivatives on supports within fixed beds or fluidized beds. Liquid-phase hydrogenation predominantly employs metals with extensive surface areas, such as Raney nickel, Raney nickel–iron, Raney cobalt, and Raney copper, due to their relatively lower cost.
Precious-metal catalysts, such as Pt and Pd, are generally used at concentrations of 0.5–5 wt % on high-surface-area support materials like charcoal, silica, aluminum oxide, or alkaline-earth carbonates.
The powdered catalyst form is suitable for slurries, while pellet forms are utilized in fixed beds. It’s imperative to optimize conditions that allow catalyst reuse and regeneration, as well as being economical for large-scale (>500 t/a) production.
Decomposition and oxidation byproducts frequently lead to catalyst deactivation by surface blockage. Even small quantities of sulfur, arsenic, or antimony compounds can poison catalysts. However, these catalyst poisons exhibit less impact as oxidation states increase (e.g., As3- → As5+ or S2- → S4+).
With noble metal catalysts, halide, sodium, magnesium, cobalt, and CO2 ions can reduce activity. Nitro aromatic compounds containing sulfur demonstrate better reduction using molybdenum or tungsten sulfide, or with nickel catalysts containing zinc or calcium carbonate.
Pd on carbon catalysts can be affected by copper salts, while both nitrophenol and nitrocresol act as strong catalyst poisons and decomposition activators during the hydrogenation of dinitrotoluene.
Hydrogenation of Halide-Substituted Nitro Compounds
The hydrogenation of nitro compounds with halide substitutions often necessitates lower temperatures to prevent halide-hydrogen exchange. Catalysts, particularly sulfide-poisoned metal catalysts, exhibit reduced activity under such conditions.
1-Chloro-2-nitrobenzene can be effectively reduced at 80 °C and 1.0–5.0 MPa (10–50 bar) using a 5 wt % Pt on carbon catalyst treated with dilute sulfuric acid, followed by H2 and H2S. This catalyst can be recycled up to 30 times without additional purification.
Similar methodologies have been employed for the hydrogenation of 3-chloronitrobenzene using catalysts treated with thiocyanate. The use of modified catalysts enhances the selectivity and effectiveness of the reaction, resulting in high yields and excellent product purity.
Equipment and Construction Materials
Hydrogenation in the vapor phase is typically conducted in continuous mode, whereas liquid-phase hydrogenation can be achieved by batch or continuous processes, depending on the scale. Continuous liquid-phase hydrogenation is primarily employed for a select few large-scale products, such as dinitrotoluene, nitrotoluene, and 1-nitronaphthalene.
However, the majority of aromatic amine production involves batch hydrogenation using catalyst slurries. This approach allows for swift transitions between products, a luxury not afforded when using fixed-bed catalysts.
Effective mixing of the three phases (hydrogen gas, nitro compound solution, and solid catalyst) is important. Rapid transport of reactants to active catalyst centers and subsequent product removal are vital for optimal reactions.
While traditional batch catalytic hydrogenations use stirred steel or stainless steel autoclaves, the growing adoption of loop reactor technology has led to improved performance. Loop reactors have been successfully employed for catalytic hydrogenation of aromatic nitro compounds by Buss (Basel, Switzerland).
The advantages over stirred autoclaves include enhanced heat and mass transfer, as well as improved reaction selectivity. Continuous hydrogenation generally results in shorter batch cycle times and increased product yields. Also, catalyst consumption is often reduced.
Loop reactors offer a straightforward and reliable method for transitioning from pilot plant to production, as scaling up is primarily determined by nozzle configuration and pump-around rate.
In contrast, scale-up for stirred autoclaves is more complex due to the significant discrepancy between small pilot vessels and large production vessels (e.g., 1:1500 to 1:12500 ratios).
Temperature and concentration gradients, along with longer reaction times in larger production vessels, often lead to unintended side reactions and reduced product yields.
Loop reactors mitigate these challenges by confining reactions to a highly agitated, well-defined zone with substantially shorter cycle times.
2. Production of Aromatic Amines by Nucleophilic Substitution
Generally all non-carbon substituents present on the aromatic ring can be replaced by amino groups but only a select few of these substitution reactions hold significant industrial significance. These include:
- Substitution of Halide Groups
- Substitution of Hydroxyl and Ether Groups
- Substitution of Sulfo Groups
2.1. Exchange of Halide
Halide substitution is useful in generating amines containing mono-, poly-, or heterocyclic substituents, along with diarylamines. Additionally, this process extends to arylpolyamines, yielding numerous products of relevance, particularly as intermediates for dyes and pigments.
2.1.1. Activated Halides
Aromatic halides that contain ortho and/or para electron-withdrawing groups, such as -NO2 or -CN, can undergo nucleophilic aromatic substitution reactions. Both o-chloronitrobenzene and p-chloronitrobenzene undergo reactions with ammonia at 170 °C, while 2,4-dinitrochlorobenzene react at a lower temperature of 70 °C without requiring a catalyst.
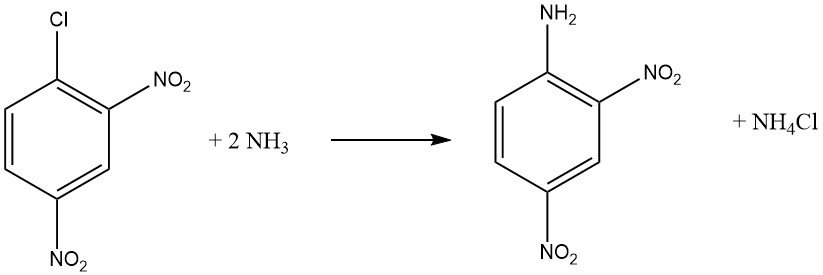
The rate of reaction between the haloaromatic compound and the amine is influenced by the basicity of the amine, except when steric hindrance is a factor.
When reacting methylamine or ammonia with a solution or suspension of 2,4-dinitrochlorobenzene at higher temperatures, N-methyl-2,4-dinitroniline is obtained in good yields, instead of 2,4-dinitroaniline.
Reaction Conditions:
The synthesis of 4-nitroaniline involves reacting 4-chloronitrobenzene with an excess of aqueous ammonia in an agitated titanium reactor at 175 °C and 4.2 MPa (42 bar) for a duration of 10 hours, yielding a 99.3% yield.
Another example is the preparation of 4-(4-methylanilino)-3-nitrobenzenesulfonamide, achieved by heating 4-chloro-3-nitrobenzenesulfonamide with p-toluidine at 130 °C for 5 hours, resulting in a 90.1% yield.
In the synthesis of 4-nitrodiphenylamine, a precursor for rubber antiozonants, 4-nitrochlorobenzene is condensed with aniline in chlorobenzene. However, N-alkylated p-phenylenediamine derivatives cannot be produced in an aqueous medium.
2.1.2. Unactivated Halides
Unactivated halides exhibit elimination-addition reactions via a benzyne intermediate, a process demanding higher temperatures than nucleophilic aromatic substitution. Such reactions yield isomeric mixtures if additional substituents are present, limiting their industrial application.
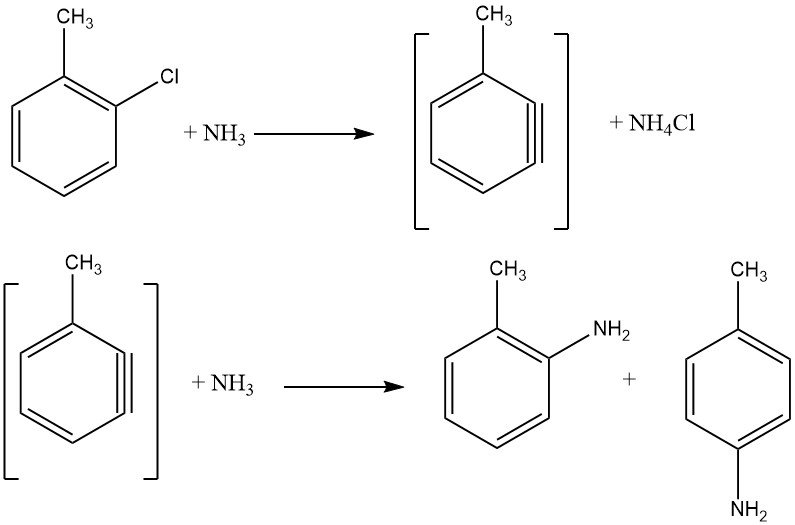
The reaction between chlorobenzene and ammonia occurs at reasonable rates only above 200 °C, often necessitating the presence of a catalyst like copper(I) and copper(II) salts or their amine complexes. Metal amides, such as NaNH2 and LiNEt2, are also employed. The energy required to facilitate halide exchange is inversely proportional to the order of activated halides: F < Cl < Br < I.
Reaction Conditions:
Typically conducted in an aqueous suspension or solution at elevated temperatures and pressures, careful temperature monitoring is crucial during the heating phase in batch processes due to the exothermic nature of arylhalide-amine reactions.
Continuous processes generally mitigate this concern. The released hydrogen halide is neutralized using excess ammonia or by adding bases like sodium carbonate or calcium oxide. With a substantial excess of amine, the reaction usually proceeds with near-complete yield.
In cases of insufficient excess of strongly basic amines in aqueous solutions, side reactions like hydrolysis or diarylamine formation may occur.
The hydrolysis of cyanogroups to amides in aqueous solutions can be averted by using inert solvents. Carboxylic acids can be readily transformed into the corresponding amides by conducting the reaction in nonaqueous media.
Materials of Construction:
At elevated temperatures and pressures, the presence of halides and amines can induce severe corrosion, making material and equipment selection a challenge. While normal steel autoclaves may suffice for laboratory purposes, substantial wall thickness reduction is to be expected (a few millimeters per year).
Additionally, stress corrosion can hasten reactor deterioration. Low-alloy steels are ill-suited for production units, particularly those employing continuous reactors. Successful reactions in aqueous media have been achieved using stainless steels, specifically chromium- and nickel-containing austenitic steels of the 316 types.
However, even these materials are prone to pitting, leading to the utilization of more corrosion-resistant alloys like Hastelloy and Inconel. Reactor materials like zirconium, titanium, and tantaclad (steel explosion clad with tantalum) have also found applications.
2.2. Exchange of Hydroxyl and Ether Groups
The displacement of an aromatic hydroxyl group by an amine is not confined to phenols and cresols; it extends to quinoline and isoquinoline derivatives as well. Ammonia can substitute activated alkoxy groups in ortho or para positions, where activation is induced by nitro, halo, or cyano groups.
For exemple, 2,4-dinitroanisole reacts with ammonia at temperatures ranging from 50 to 200 °C, yielding 2,4-dinitroaniline with a yield of 50 to 90%.
Interest in phenol-amine reactions often hinges on the cost of phenol raw material. The primary exchange processes are:
1. Aqueous medium reaction without a catalyst:
At elevated temperature and pressure, 3-nitrosalicylic acid reacts with aqueous ammonia to yield 2-amino-3-nitrobenzoic acid.
2. Reaction in the presence of an acid catalyst:
m-Aminophenols form by the reaction of resorcinol and aqueous ammonium hydroxide or alkylamines, with boric acid as the catalyst.
By employing the same catalyst under anhydrous conditions, resorcinol reacts with ethanolamine or 3-amino-1-propanol to yield 3-(β- and γ-hydroxyalkylamino)phenols. An analogous process utilizes an acid-activated alumina-silica catalyst.
3. Multiphase system reactions yield nitroanilines from nitrophenols and ammonia, preferably by employing a solvent where the reaction product has good solubility. Numerous phase-transfer catalysts are being employed in these reactions.
4. p-Nitrosoaniline and p-nitroso-N-phenylamine are synthesized in liquid ammonia using either an ammonium salt or a tertiary amine in an organic solvent.
5. p-Nitrosophenols undergo reactions with alkylamines through p-nitrosophenyl ethers, yielding the corresponding N-substituted p-nitrosoanilines. The water produced in the process is removed to drive the reaction to completion.
6. The Halcon process is a significant vapor-phase amination of phenol involving the reaction of phenols or naphthols with ammonia in the gas phase at 250 °C using metallic or non-metallic oxides, such as MgO, B2O3, Al2O3, SiO2, TiO2, or mixtures.

U.S. Steel Chemicals has employed the Halcon process since 1982 at its Haverhill, Ohio plant, producing aniline from phenol (aniline capacity: 100,000 t/a).
7. The Bucherer reaction is the reaction of 1- or 2-naphthols with aqueous amine or ammonium solutions using hydrogen sulfite as a catalyst. This reaction is primarily used for the naphthalene series. The use of hydrogen sulfite reduces the reaction temperature by 50 to 150 °C and yields high purity and yield (85 – 95%).
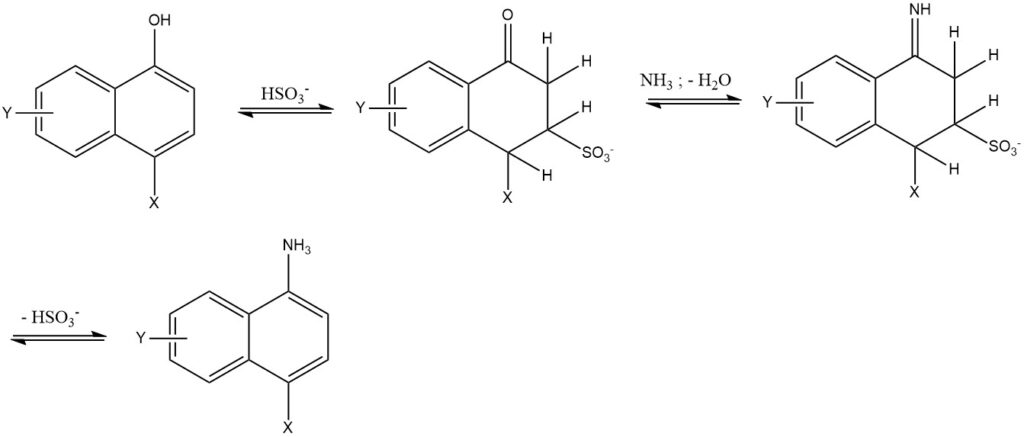
2.3. Exchange of Sulfo Groups
Ammonia has the capacity to substitute sulfo groups in benzene, naphthalene, and anthraquinone derivatives. However, this exchange process holds significant industrial importance only within the anthraquinone series.
In the past, the production of 1-aminoanthraquinones involved the reaction of 1-sulfoanthraquinones with amines. To counteract the release of hydrogen sulfite, stoichiometric quantities of an oxidizing agent, such as sodium 3-nitrobenzenesulfonate, were introduced.
A more recent approach for synthesizing 1-aminoanthraquinone involves the reduction of the corresponding nitroanthraquinone. This methodology eliminates the need for the mercury-catalyzed sulfonation leading to 1-sulfoanthraquinone.
By a process conducted in an aqueous medium under pressure at temperatures ranging from 100 to 200 °C, 1-cyclohexylaminoanthraquinone-5-sulfonic acid is generated from anthraquinone-1,5-disulfonic acid and cyclohexylamine. The oxidation of hydrogen sulfite is achieved using substances like sodium perchlorate or 3-nitrobenzenesulfonic acid.
3. Other Processes
Various methods outlined in the literature for aromatic amine synthesis will be highlighted in this discussion. It is important to note that only a selection of these methods will be covered here.
The large-scale production of 4,4′-methylenedianiline involves the condensation of aniline with formaldehyde in either aqueous or aqueous-methanolic solutions.
Diphenylamine is synthesized by vapor-phase condensation of aniline over alumina or titanium catalyst at temperatures ranging from 450 to 500 °C. Alternatively, it can be produced in the liquid phase at temperatures from 175 to 450 °C.
Arylhydroxylamines can be rearranged in dilute sulfuric acid to form o- and p-aminophenols.
An important pigment intermediate, 3,3′-dichlorobenzidine, is manufactured by Bofors (located in Muskegan, MI). The process involves the benzidine rearrangement initiated from o-chloronitrobenzene.
Reference
- Amines, Aromatic; Ullmann’s Encyclopedia of Industrial Chemistry. – https://onlinelibrary.wiley.com/doi/10.1002/14356007.a02_037
-
Process for the production of aromatic amines. – https://patents.google.com/patent/US5877350A/en