
Nitration of Aromatic Compounds
1. Unraveling the History of Aromatic Nitration Reactions
During the early 19th century, significant advancements were made in the synthesis of aromatic nitro compounds. Mitscherlich in 1834 successfully obtained such compounds by subjecting hydrocarbons derived from coal tar to fuming nitric acid.
Laurent continued this work in 1835, focusing on the nitration of naphthalene, the most readily available pure aromatic hydrocarbon at that time. In 1838, Dale presented findings on mixed nitro compounds derived from crude benzene during the annual meeting of the British Association for the Advancement of Science.
However, it wasn’t until 1845 that Hofmann and Muspratt reported their systematic research on the nitration of benzene, resulting in mono- and dinitrobenzenes using a mixture of nitric and sulfuric acids.
Table of Contents
The initial production of nitrobenzene was carried out on a small scale and carefully distilled to yield a yellow liquid with a bitter almond-like aroma. This product, known as ‘essence of mirbane,’ was sold to soap and perfume manufacturers.
Bechamp’s iron reduction process, which increased the availability of aniline, was published in 1854. Subsequently, Perkin’s discovery of aniline mauve in 1856 marked the beginning of the European aniline dye industry, serving as the foundation for the worldwide synthetic colorant industry, with estimated sales of $6 x 109 in 1988.
The development and scale-up of the nitration and reduction processes, initiated by Perkin and continued by numerous others, significantly reduced the trade price of aniline from 50 Fr/kg in 1858 to 10 Fr/kg in 1863.
By 1871, European aniline production had reached 3500 t/a. In 1985, European aniline production rose to 500,000 t/a, with its use in colorants accounting for only 4% of overall aniline production.
The majority of growth was attributed to rubber chemicals and isocyanates, consuming over 50% of nitrobenzene production for methylene diphenyldiisocyanate manufacturing.
Naturally occurring nitroaromatic compounds are limited in number, with chloramphenicol being the first recognized in 1949, extracted from cultures of Streptomyces venezuelae, a soil mold.
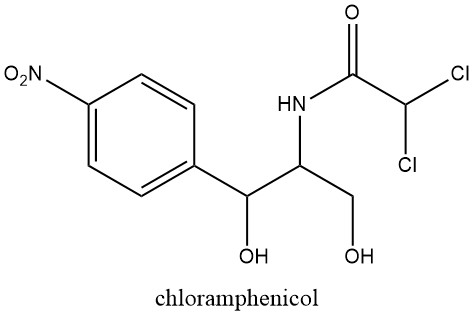
This discovery prompted further investigations into the pharmacological activity of the nitro group, following the earlier discovery of the antibacterial properties of nitrofuran derivatives in 1943.
Many synthetic pharmaceuticals and agrochemicals also contain nitroaromatic groups, although the precise role of the nitro group in these compounds often remains unclear.
The primary method for introducing nitro groups into aromatic systems is nitration.
While there are some indirect methods available, such as oxidation of nitroso or amino compounds, replacement of diazonium groups (Sandmeyer reaction), rearrangement of nitramines, and nucleophilic displacement reactions, they have limited industrial significance.
Unit processes, like sulfonation, halogenation, or amination, are commonly applied to primary nitro starting materials, mainly derived from nitrobenzene, nitrotoluenes, and nitrochlorobenzenes, to produce various important nitro compounds.
Nucleophilic displacement of activated nitro groups has some synthetic potential, and its nucleofugicity may be comparable to that of fluorine substituents. Certain readily available nitro compounds, like 1,3-dinitrobenzene, react with specific reagents to form useful derivatives.
Nitro compounds or their derivatives serve as intermediates for colorants, agrochemicals, pharmaceuticals, fine chemicals, synthetic materials, and explosives, with few major outlets for large-scale production.
2. Nitration Reaction
The process of nitration can be described as the irreversible introduction of one or more nitro (NO2) groups into an aromatic nucleus, replacing a hydrogen atom.
Nitration is an electrophilic substitution reaction, represented by the equation:
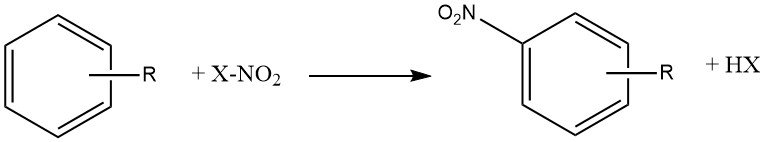
The introduction of the nitro group deactivates the ring towards further electrophilic substitution, making dinitration a rare occurrence under the conditions used for mononitration.
More vigorous conditions are usually required for dinitration, necessitating excess, stronger acid, and higher temperature. These conditions are often applied to isolated mononitro compounds, rather than carrying out stepwise reactions in situ.
The nitration reaction is highly exothermic, as evidenced by the mononitration of benzene (ΔH = -117 kJ/mol) and naphthalene (ΔH = -209 kJ/mol). Consequently, it is one of the most potentially hazardous industrially operated unit processes.
The heat generated during nitration can cause nitric acid to exothermically degrade organic materials into gaseous products with explosive violence.
Nitroaryl compounds, especially those with multiple nitro groups, pose potential hazards due to their high oxygen contents.
Some polynitro compounds, like trinitrotoluene and picric acid, are detonable and have long histories of use as explosives. Additionally, nitroaromatic compounds may undergo violent decomposition reactions when heated with alkali.
The nitration process can be conducted using either a batch (discontinuous) or continuous basis. Batch reactions are suitable for lower tonnage requirements and are performed in nitrators designed to handle various products.
For safety reasons, these reactors are typically limited to about 6000-L capacity and equipped with efficient agitators and cooling systems.
On the other hand, continuous reaction, suitable for large-tonnage intermediates like nitrobenzene and nitrotoluenes, offers advantages in terms of safety and cost efficiency.
Nitroaromatic production primarily relies on high-yield processes, with over 80% of the total cost attributed to raw materials. Therefore, operational savings are limited.
However, improvements have been made in areas such as sulfuric acid recycling, an integral requirement in efficient nitration processes, and isomer control and separation, contributing to enhanced process efficiency.
3. Nitrating Agents
For practical and economic reasons, industrial-scale nitration is commonly carried out using a mixed acid, which is a combination of nitric and sulfuric acids. Occasionally, other combinations, such as aqueous nitric acid, nitric acid in acetic acid, or nitric acid in acetic anhydride, are utilized.
In certain important supporting studies, alternative component acids like perchloric acid, hydrofluoric acid, or boron trifluoride may be used, but they are limited in their application. These alternative reactions may take place in inert organic solvents like chlorohydrocarbons or sulfolane to create homogeneous reaction mixtures.
The nitrating agent’s strength (X-NO2) decreases as the electronegativity of X decreases. The most active species in these systems is the nitronium ion (NO2+). In the common system, the overall equation is a composite of various equilibria present in HNO3 – H2SO4 – H2O mixtures, which should be considered when assessing substrate reactivity and the extent of required nitration.
For large-scale aromatic mononitration, a typical nitrating agent consists of 20% nitric acid, 60% sulfuric acid, and 20% water, often referred to as “20/60/20 mixed acid.” Alternatively, in some situations, it can be described as 15 mol % nitric acid, 30 mol % sulfuric acid, and 55 mol % water.
The liquid aromatic substrate and the nitrated product typically form a separate phase from the aqueous mixed acid. Therefore, efficient agitation is crucial to maximize contact with the organic phase and minimize resistance to mass transfer.
Solid substrates are best dissolved in the sulfuric acid phase. It is essential to optimize various variables, such as nitric acid concentration, temperature, and other parameters for each specific nitration to achieve the desired isomer formation and minimize side reactions.
Free-radical reactions, typically employed in the vapor phase with nitric acid or nitrogen dioxide as the nitrating agent, are not commonly used in aromatic nitration. They are more suitable for aliphatic nitration.
4. Nitration Reaction Mechanisms
Extensive experimentation has been conducted on aromatic nitration since the confirmation of the nitronium ion (NO2+) as the active species by the Ingold-Hughes school in 1950, using Raman spectra analysis.
While temperature and degree of mixing play crucial roles in all nitrating agents, other variables often lead to inconsistent results. In commercial nitration using HNO3-H2SO4, the relative rate constants exhibit a wide range, resulting in reaction times varying from several seconds for active substrates to several hours for inactive substrates.
It has been argued that in the non-ideal conditions of large-scale production, mass transfer may have more control over nitration than kinetics, challenging the assumption in many laboratory studies that mass-transfer resistance is negligible.
Additionally, an earlier assumption that the rate of nitration aligns with the equilibrium formation of the nitronium ion was challenged due to the realization that different mechanisms may apply at varying acid strengths.
Figure 1 illustrates the mechanism that can account for all available facts on the nitration of simple substrates such as benzene and toluene (excluding phenols or polyalkylbenzenes). This mechanism involves four rate constants and three significant reversible reactions, making the kinetics highly complex, even if the reaction is entirely kinetically controlled.
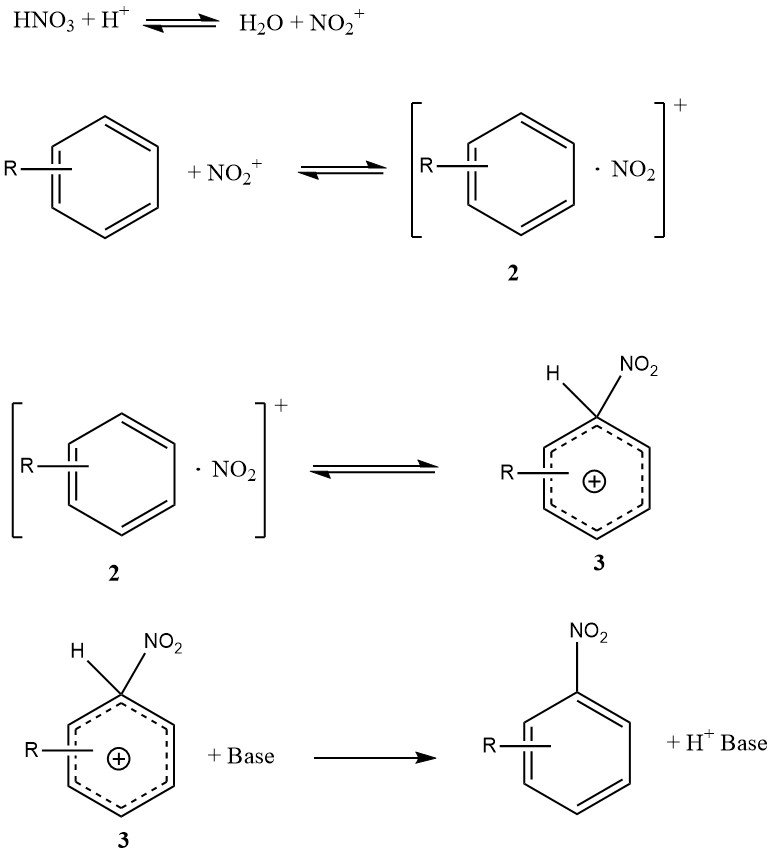
The encounter complex or encounter pair (2) is a novel feature of this mechanism. It is a molecular complex of a less specific nature compared to the earlier concept of an intermediate p-complex. Importantly, the encounter complex (2) plays a kinetic role and may indeed be rate-determining.
The conversion of the s-complex (3), sometimes referred to as a Wheland intermediate, to the product is not rate-limiting because the nitration reaction does not exhibit a primary kinetic isotope effect.
Under specific conditions, the formation of the nitronium ion might be rate-determining, but for reactive substrates in aqueous sulfuric acid, the formation of the encounter complex (2) is predominantly the rate-determining step.
In contrast, the nitration of toluene, benzene, and other less reactive aromatic compounds follows an overall rate law where the formation of the s-complex (3) is mainly the rate-determining step.
In the case of phenol, when the ring is highly activated towards electrophilic attack, the nitrosonium ion (NO+) becomes significant and leads to the formation of an intermediate nitroso compound. This intermediate is further oxidized by nitric acid to yield the nitro derivative while regenerating nitrous acid.
Another exception to the general mechanism is ipso-electrophilic attack at a substituent position. With di- and polyalkylbenzenes, the ipso-nitroarenium ion (4) may form instead of the conventional Wheland intermediate (3).
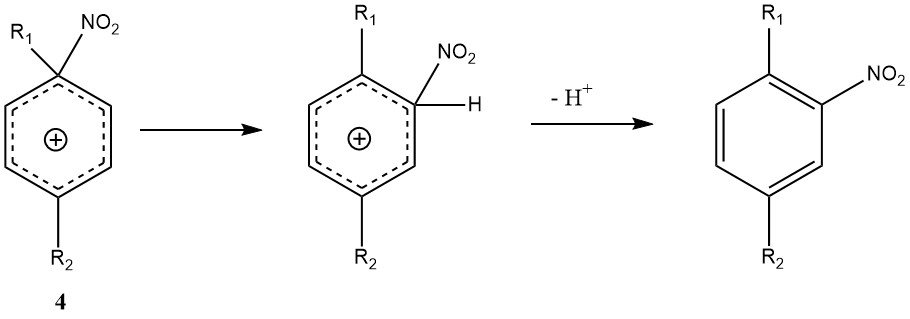
The rearrangement of 4 to the Wheland intermediate can occur, but it is accompanied by side reactions such as nitro-dealkylation, making the nitration of polyalkylbenzenes potentially yielding more variable mixture products than other nitrations.
Improved understanding of the formation of ipso-Wheland intermediates has led to their utilization in other reactions, particularly with nucleophiles.
Reference
- Nitro Compounds, Aromatic; Ullmann’s Encyclopedia of Industrial Chemistry. – https://onlinelibrary.wiley.com/doi/10.1002/14356007.a17_411
FAQ
The nitration reaction is a chemical process in which one or more nitro (NO2) groups are introduced into an aromatic nucleus by replacing a hydrogen atom. It is an electrophilic substitution reaction commonly used to modify aromatic compounds by attaching nitro groups, which can significantly alter their properties and reactivity.
The primary chemicals used in nitration are nitric acid (HNO3) and sulfuric acid (H2SO4), which together form a mixture known as mixed acid. Other chemicals that may be occasionally used include aqueous nitric acid, nitric acid in acetic acid, or nitric acid in acetic anhydride. Perchloric acid, hydrofluoric acid, or boron trifluoride may be utilized in specific supporting studies.
In aromatic nitration, the electrophile responsible for the attack on the aromatic nucleus is the nitronium ion (NO2+). The nitronium ion is formed by the reaction between nitric acid and sulfuric acid and acts as the electrophile, accepting a pair of electrons from the aromatic ring during the electrophilic substitution reaction.
Sulfuric acid in aromatic nitration acts as a catalyst in the formation of the nitronium ion (NO2+) from the reaction between nitric acid and sulfuric acid. Additionally, sulfuric acid helps in dehydrating the nitric acid, making it a stronger nitrating agent. Furthermore, sulfuric acid helps to dissolve and solubilize the aromatic substrate, enabling efficient reaction and maximizing contact with the nitrating agent.
The primary products of nitration are mono-substituted nitroaromatic compounds, which are formed when one nitro group is introduced into the aromatic nucleus. Five potential products from nitration can include:
1. Nitrobenzene (C6H5NO2)
2. Nitrotoluenes (e.g., ortho-nitrotoluene, meta-nitrotoluene, and para-nitrotoluene)
3. Nitronaphthalenes (e.g., alpha-nitronaphthalene and beta-nitronaphthalene)
4. Nitrophenols (e.g., ortho-nitrophenol, meta-nitrophenol, and para-nitrophenol)
5. Nitroanilines (e.g., ortho-nitroaniline, meta-nitroaniline, and para-nitroaniline)